
Abstract
The clinical signs and pathology of the central nervous system in 9 horses with naturally occurring neurologic disease due to Trypanosoma evansi are described. The clinical course was 2 to 20 days; clinical signs included marked ataxia, blindness, head tilt and circling, hyperexcitability, obtundity, proprioceptive deficits, head pressing, and paddling movements. Grossly, asymmetric leukoencephalomalacia with yellowish discoloration of white matter and flattening of the gyri were observed in the brain of 7 of 9 horses. Histologically, all 9 horses had necrotizing encephalitis that was most severe in the white matter, with edema, demyelination, and lymphoplasmacytic perivascular cuffs. Mild to moderate meningitis or meningomyelitis was observed in the spinal cord of 5 of 7 horses. T. evansi was detected immunohistochemically in the perivascular spaces and neuropil of formalin-fixed, paraffin-embedded brain tissue in 8 of 9 horses.
Keywords
Trypanosoma evansi is a flagellate protozoan parasite that causes disease in several mammalian species. The hind limb weakness of equine trypanosomiasis gave rise to the disease name mal das cadeiras in Brazil or mal de caderas in the Spanish-speaking countries of South America. Both terms translate as “sickness of the hips.” The disease is called murrina in Panama and surra in many Asian countries. 16, 22 In the past, T. evansi was designated as T. equinum, T. hippicum, and T. venezuelense. 16 In South America, capybaras (Hydrochoerus hydrochaeris), 15, 30, 33 coatis (Nasua nasua), 32 small marsupials (e.g., Monodelphis spp.), and armadillos (Dasypus spp.) may serve as reservoirs for T. evansi. 15 Unlike the African Trypanosoma species that cause nagana in animals and “sleeping sickness” in human beings, T. evansi does not require the biologic cycle in Glossina spp. flies, and it is mechanically transmitted by biting insects, especially tabanids and stomoxids, by vampire bats (Desmodus rotundus) 16 and, possibly, by ticks. 4
Trypanosomiasis in horses is characterized by intermittent fever, anemia, progressive weakness, loss of body condition, and unstable gait. 11, 22 Neurologic signs have been described occasionally in the terminal phase of natural infection by T. evansi in horses, 40 cattle, deer, 46, 47 and buffaloes, 45 but have not been well documented in horses.
In Brazil, T. evansi is enzootic in horses from the midwestern part of the country, specifically the Pantanal region, 7, 8, 10, 15, 41– 43 where the disease has great economic importance because of the large equine population. 1, 41 Outbreaks of T. evansi infection with the death of at least 100 horses were reported for the first time in southern Brazil in 2005. 38 Twenty-three affected horses were clinically evaluated; 9 of these had severe, fatal neurologic disease. This paper describes the neurologic signs and the pathologic and immunohistochemical findings in the central nervous system of those 9 horses naturally infected by T. evansi.
Materials and Methods
The cases of spontaneous T. evansi infection in horses occurred from January 2003 to February 2004 in the western part of the state of Rio Grande do Sul in southern Brazil. Data were collected by the authors during visits to 4 stud farms of the Crioulo breed where the outbreaks occurred. 38 Clinical examinations were performed on 23 affected horses; necropsy examinations were performed on 15. All but 1 affected horse had chronic wasting with anemia; 8 of the horses had severe neurologic signs; an additional horse developed neurologic signs without wasting (Table 1). The authors did not investigate anecdotal reports of trypanosomiasis outbreaks on neighboring horse farms.
Data from 9 horses with neurologic disease associated with natural T. evansi infection.*

NR = not reported; NI = not informed.
Horse that developed only neurologic signs without wasting.
Euthanized animals.
Serologic evaluation for T. evansi by enzyme-linked immunosorbent assay was carried out in 67 horses from one of the farms. Five serum samples originated from horses with neurologic signs; the remaining 62 samples came from horses with no apparent signs that were randomly selected on this farm. Horses with antibody concentrations just above the cutoff level (optical density, 0.385) were considered to have low titers against T. evansi, whereas horses with similar or higher antibody concentrations than those of the positive control (optical density, 1.422) were considered to have high titers.
Of the 9 horses with neurologic signs, 6 died, and 3 were euthanized with an overdose of barbiturates. The horses were necropsied within 24 hours of death; the brain and spinal cord were fixed for 7 to 30 days in 10% neutral-buffered formalin and then sliced coronally at 1-cm intervals. The following regions of the brain and cord were processed routinely for histologic examination and immunohistochemistry: medulla at the level of the obex; pons and cerebellar peduncles; mesencephalon at the level of rostral colliculi; thalamus; cerebellum; occipital lobe; parietal lobe; frontal lobe; hippocampus; basal nuclei; cervical spinal cord (horse Nos. 2–8 only); thoracic spinal cord (horse Nos. 2 and 4–8 only); and lumbar spinal cord (horse Nos. 2, 4, 5, and 8 only). Histologic lesions were evaluated and graded as 1 (mild), 2 (moderate), or 3 (marked) in order to determine their distribution and severity. The following changes were graded: inflammatory infiltrate, presence of plasma cells with intracytoplasmic eosinophilic globules (Mott cells), focal gliosis, diffuse gliosis, endothelial swelling, hemosiderophages, edema of neuropil and white matter, hemorrhage, meningitis, necrosis, axonal spheroids, and Alzheimer type 2 astrocytes.
For immunohistochemistry, sections of the brain or spinal cord were applied to glass
slides coated with poly-
Results
The details of the epidemiologic, clinical, and gross findings, and portions of the histopathologic changes have been published 38 and will only be summarized here. During the summer of 2003, approximately 200 horses on 3 bordering farms, as well as 10 mares from a fourth farm that were sent to 1 of the 3 farms for breeding, were maintained near the banks of a river where, according to farm employees, many capybaras (H. hydrochaeris) had died. Approximately 1 month after their arrival in this area, many of the horses presented with marked weight loss, muscle atrophy, and incoordination. Within 2 to 3 months, and extending through the rest of the year and into 2004, approximately 100 horses died. Capybaras were incriminated as reservoirs of T. evansi in these outbreaks. There was also anecdotal evidence that several horses on one farm had been treated with a common needle early in the outbreak.
Twenty-three horses were evaluated clinically. Twenty-two horses presented with chronic wasting. In this group the onset of disease was insidious and characterized by progressive loss of weight (despite voracious appetite), lethargy, incoordination, gait instability (wobbling) involving the pelvic limbs, atrophy of heavy muscles of the hindquarters, difficulty in rising, muscle weakness, pallor of mucous membranes, subcutaneous edema of the ventral portions of the trunk and limbs, and abortions. Three horses of this group survived but remained in poor nutritional and muscular condition.
Eight horses had neurologic clinical signs in addition to those signs mentioned above. Neurologic signs included marked ataxia (Fig. 1), blindness, circling, hyperexcitability, depression, proprioceptive deficits, head tilt, and paddling movements. An additional horse developed neurologic signs without evidence of wasting. The clinical course of chronic wasting lasted 1 to 20 months; the neurologic disease, 2 to 20 days; all cases were fatal (Table 1).

Horse No. 6, naturally infected by T. evansi. Note abnormal placement of the limbs.
Hematologic findings in affected horses included normocytic normochromic anemia, with leukocytosis due to lymphocytosis. Erythrophagocytosis and protozoa identified as T. evansi (Fig. 2) were seen in the peripheral blood of 1 horse that developed neurologic signs and in 3 other horses that developed only chronic wasting. High titers against T. evansi were detected in the sera of 4 of 5 horses with neurologic signs; titers varied from low to high in 10 of 62 serum samples collected randomly from clinically normal horses from one of the affected farms.
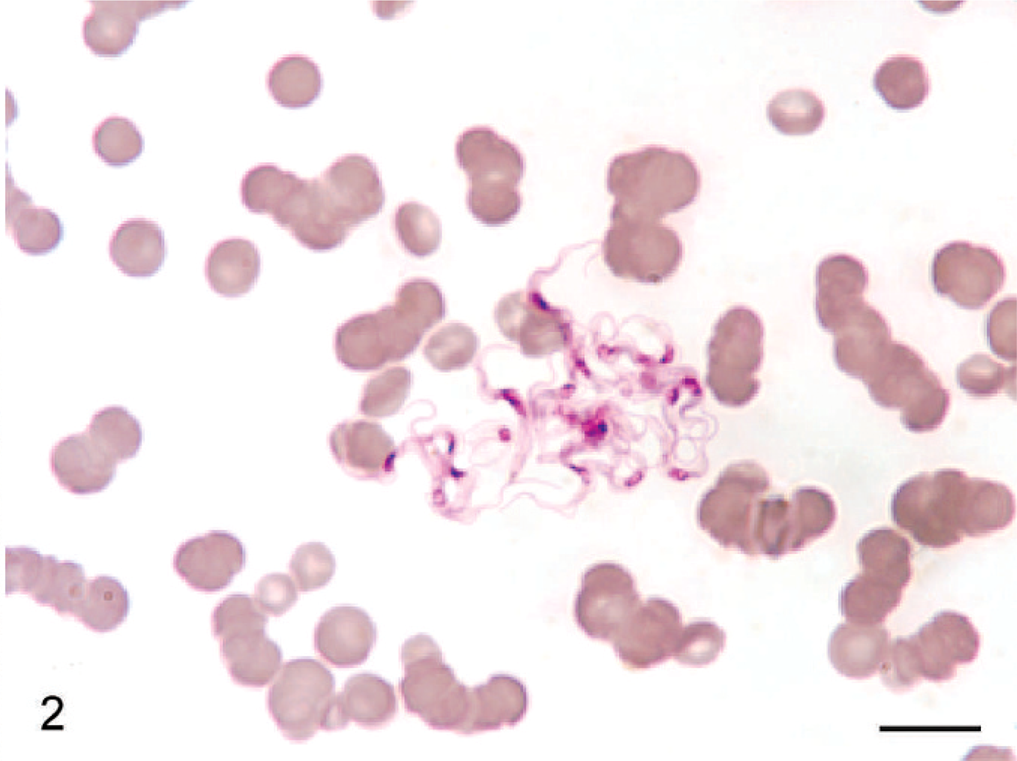
Blood smear; horse No. 2. Cluster of T. evansi trypomastigotes in the peripheral blood. Diff-quick. Bar = 10 µm.
Gross findings in horses with the chronic wasting form of the disease included marked hindquarter muscle atrophy, splenomegaly, and lymphadenomegaly. In 7 of 9 horses with neurologic clinical signs (horse Nos. 1 and 4–9), gross lesions in the brain included asymmetric swelling of the cerebral hemispheres with flattening of gyri. The white matter of the parietal, temporal, and frontal lobes was unilaterally yellow, gelatinous, and friable because of severe edema and malacia. In horse No. 8, similar lesions were observed in the basal nuclei, thalamus, and mesencephalon (Fig. 3). Moderate subpial hemorrhages were observed in the brains of horse Nos. 3 and 4.
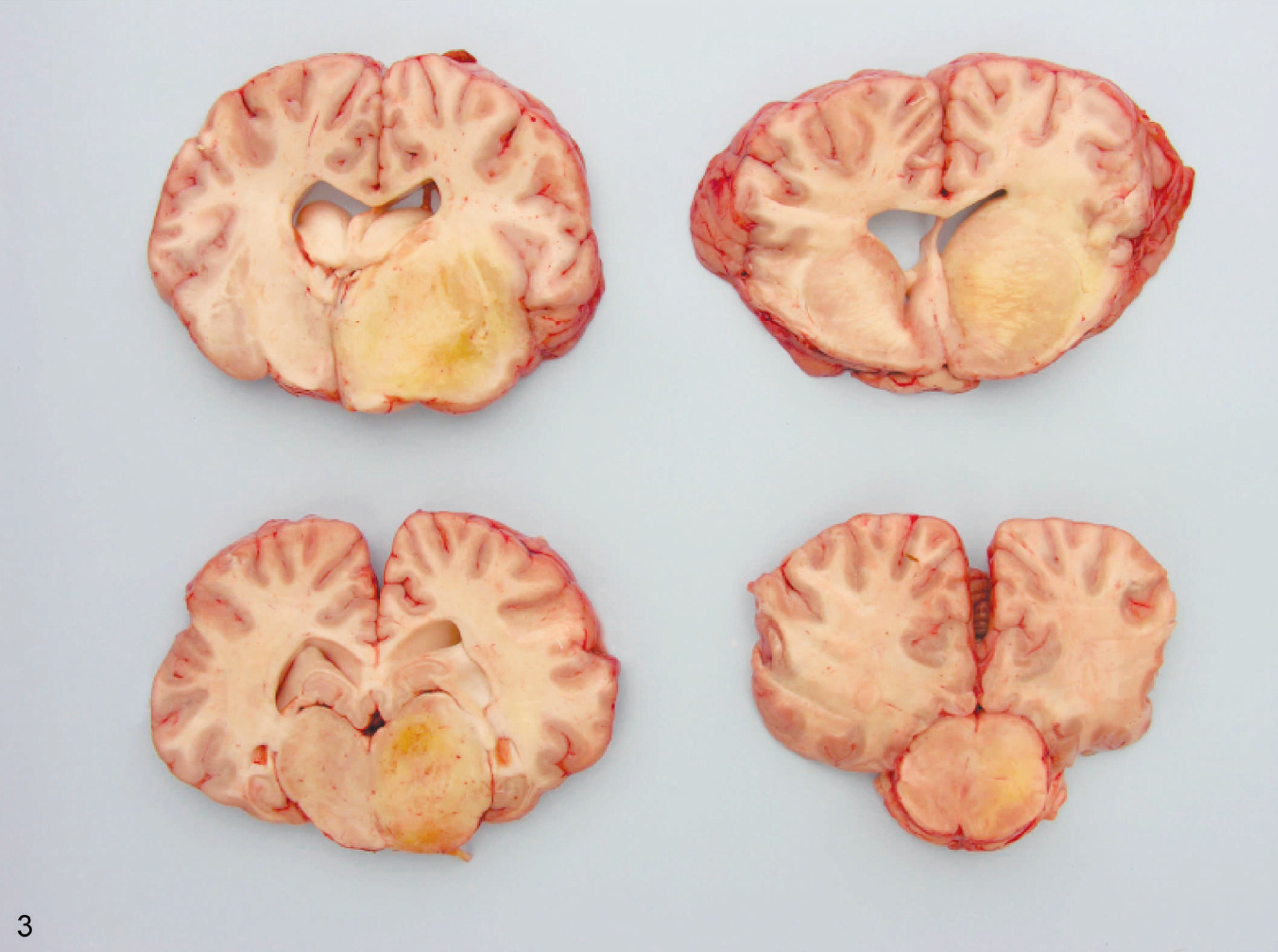
Coronal sections of brain; horse No. 8. Note asymmetry due to swelling of the right cerebral hemisphere and brain stem. Thalamus, basal nuclei, and mesencephalon have yellow, gelatinous, and friable areas of edema and malacia.
Histologically, lymphoid organs had marked follicular hyperplasia, erythrophagocytosis, and hemosiderosis. In the liver there was moderate lymphoplasmacytic periportal hepatitis, Kupffer cell hypertrophy, and hemosiderosis. Two horses had mild lymphoplasmacytic and histiocytic perineuritis with Wallerian degeneration in peripheral nerves.
The distribution and severity of lesions in the brain varied within a case and among different horses. The histologic lesions affected mainly the white matter and were characterized by moderate to severe perivascular lymphoplasmacytic meningoencephalitis (Figs. 4, 5), necrosis, edema (Fig. 6), and hemorrhage. Lymphocytes and plasma cells, often with intracytoplasmic Russell bodies (Mott cells), greatly expanded the Virchow-Robin spaces and extended into the surrounding neuropil (Fig. 7). Other features included lymphoplasmacytic karyorrhexis, infiltration by macrophages with foamy cytoplasm, focal and diffuse gliosis, scattered Alzheimer type 2 astrocytes, axonal spheroids, acute perivascular hemorrhage, and hemosiderosis. The following areas of the brain were mainly affected with decreasing severity: parietal lobe, basal nuclei region, thalamus, occipital lobe, frontal lobe, hippocampus, mesencephalon, cerebellar peduncles, cerebellum, and obex. In horse Nos. 1, 4, 7, and 9, the lesions were more severe in the right cerebral hemisphere; in horse Nos. 6 and 8, in the left cerebral hemisphere.
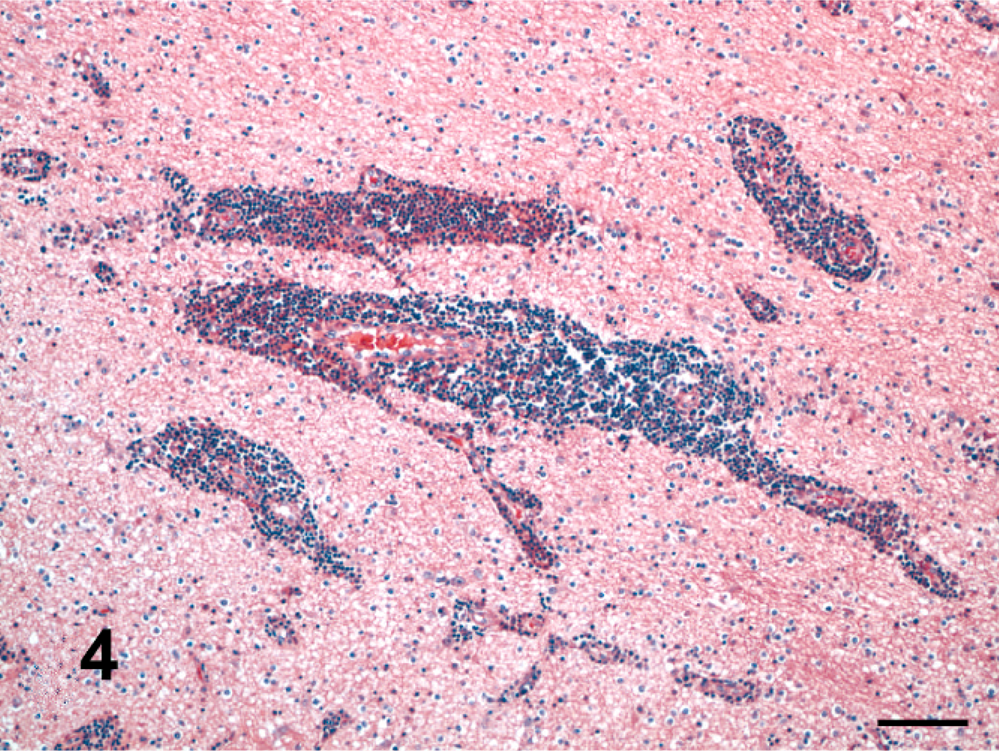
Brain, pons; horse No. 1, naturally infected by T. evansi. Marked lymphoplasmacytic infiltration of Virchow-Robin space. HE. Bar = 100 µm.
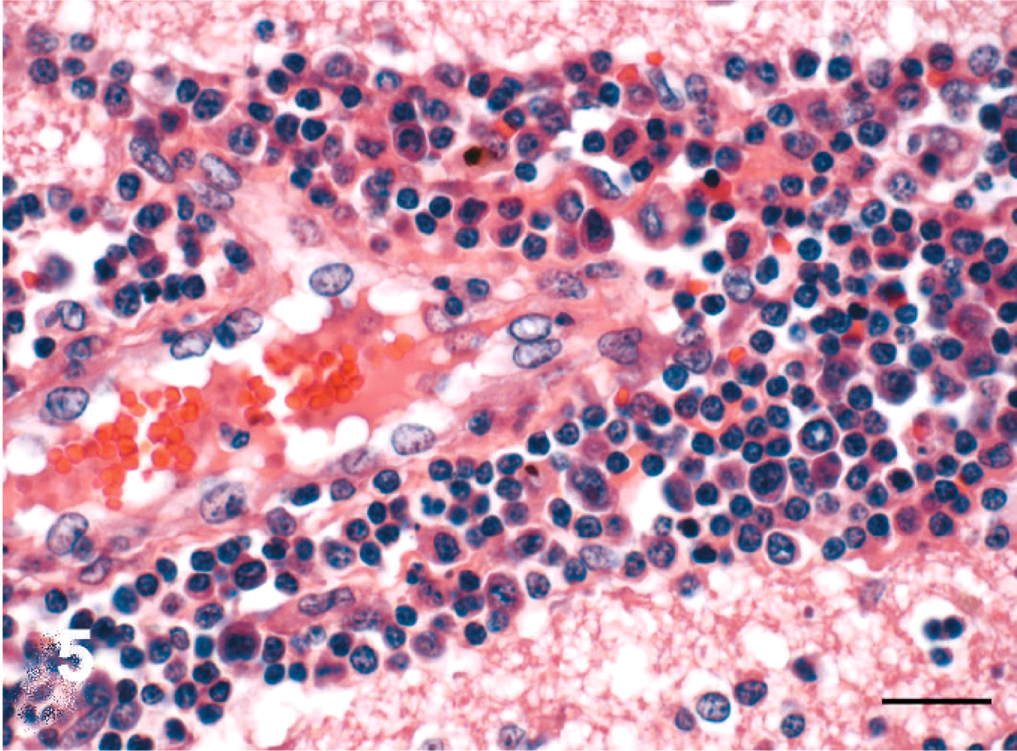
Brain, pons; horse No. 1, naturally infected by T. evansi. High-magnification view of perivascular inflammatory infiltrate. Numerous lymphocytes and plasma cells expand the Virchow-Robin space. Endothelial cells are hypertrophied. HE. Bar = 20 µm.
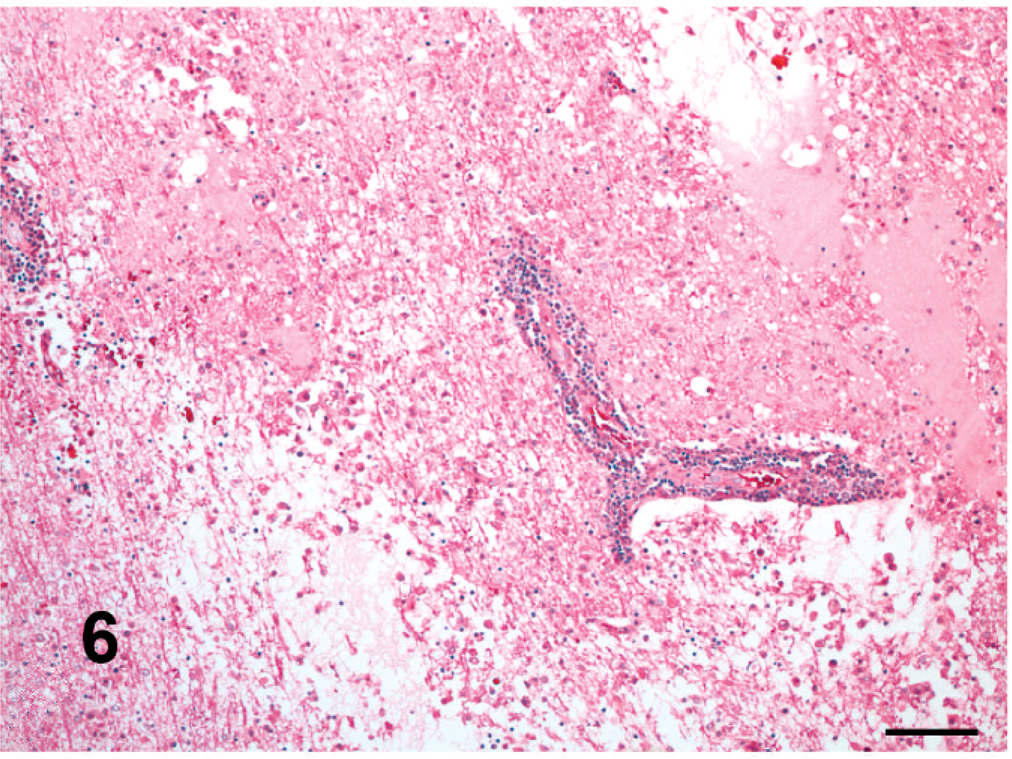
Brain, parietal lobe; horse No. 3, naturally infected by T. evansi. Areas of rarefaction of the subcortical white matter with proteinaceous edema, cavitation, and infiltration by macrophages. These areas corresponded to the gelatinous areas of yellow discoloration observed grossly. HE. Bar = 100 µm.
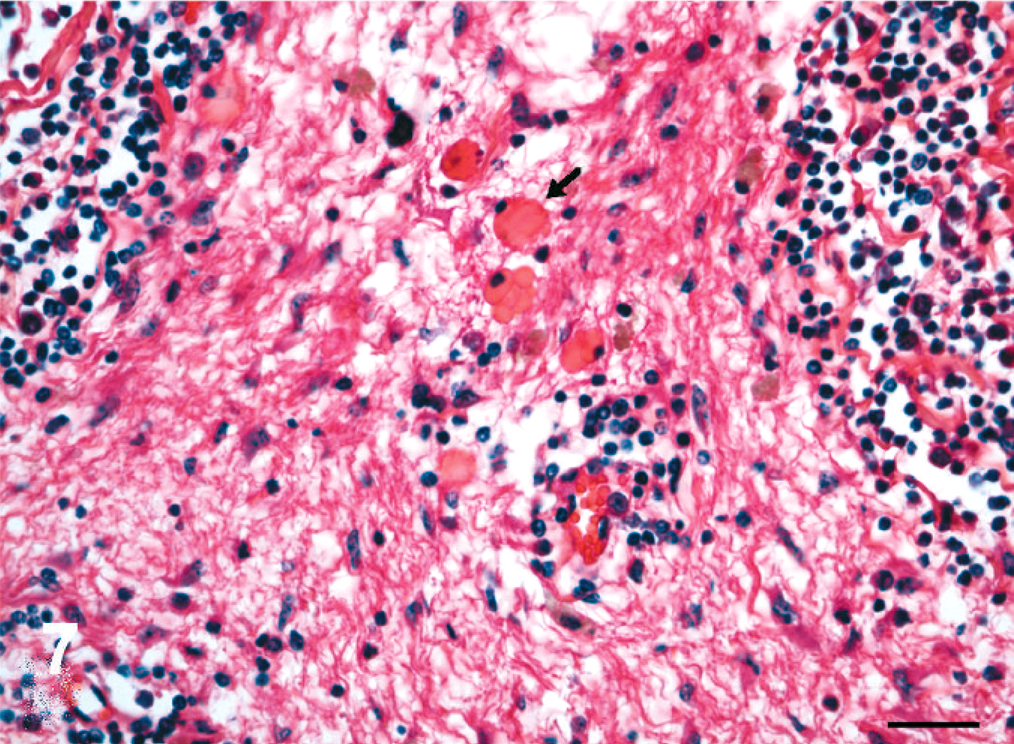
Brain, basal nuclei; horse No. 9, naturally infected by T. evansi. Mott cells with Russell bodies (arrow) in the white matter. HE. Bar = 25 µm.
No lesions were detected in the trigeminal ganglia or hypophysis. Lesions in the spinal cord (examined in 7 horses) consisted of mild lymphoplasmacytic meningomyelitis in 3 horses (horse Nos. 2, 4, and 9) and mild lymphoplasmacytic meningitis in 2 horses (horse Nos. 5 and 6). Lesions in the spinal cord tended to diminish in severity from cervical to lumbar segments.
Parasites were not detected in any horse in HE-stained sections of brain, but numerous T. evansi organisms or fragments of these organisms were detected in formalin-fixed, paraffin-embedded sections of the brain by immunohistochemistry in 8 horses (horse Nos. 1–6, 8, and 9). The parasites were in the Virchow-Robin spaces and in the neuropil (Fig. 8). Parasite nuclei were strongly immunoreactive; the cytoplasm was moderately reactive, and the cell membranes of the parasite were clearly labeled. The brain sample from 1 horse (horse No. 7) was immunohistochemically negative for T. evansi.
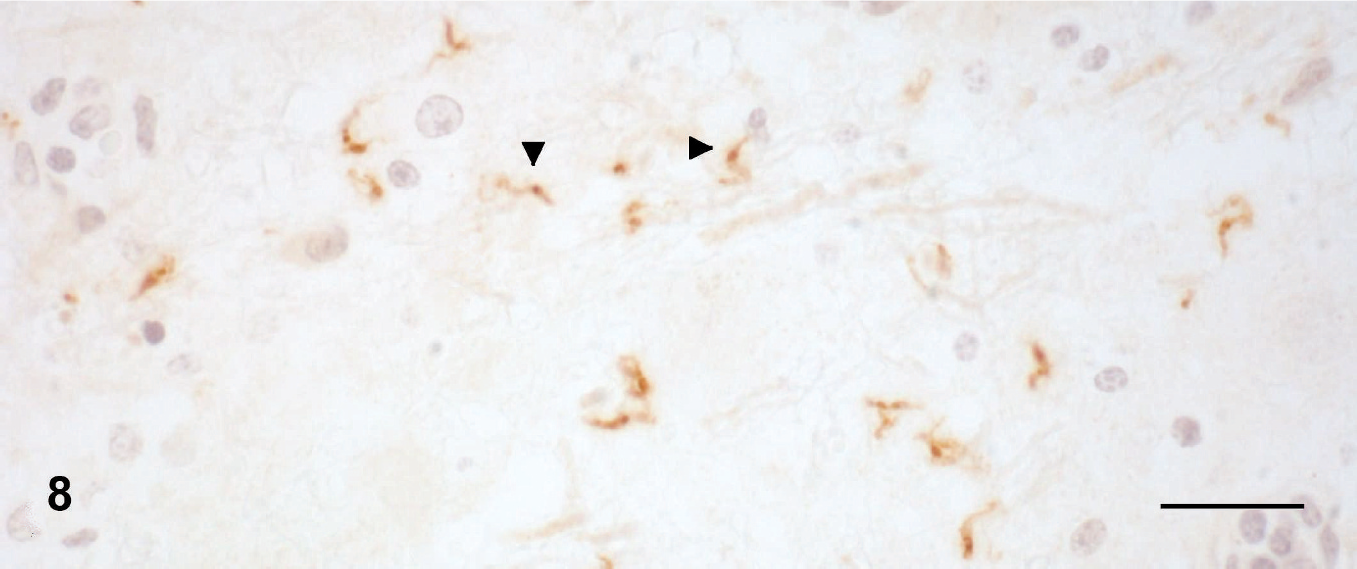
Brain, parietal cortex; horse No. 6, naturally infected by T. evansi. Trypomastigotes are labeled by T. evansi antibody (arrowheads). Immunohistochemistry. Avidin-biotin-peroxidase complex. Bar = 20 µm.
Discussion
T. evansi induces a wasting disease with a protracted clinical course 48 associated with anemia and instability (wobbling) of pelvic limbs in horses, camels, and dogs. 8, 11, 16, 41 Natural infection by T. evansi is rarely considered a cause of encephalitis in horses. 40 Neurologic signs have been reported in cattle 47 and hog deer 46 naturally infected by T. evansi; however, the histopathologic changes in the brain of these species were either not described 47 or just briefly described. 45, 46 On the other hand, mild lymphoplasmacytic meningoencephalitis was reported in horses, 21 donkeys, 3 dogs, 2 goats, 6 coatis (N. nasua), 15 and buffaloes 5 experimentally infected by T. evansi. All horses in this report that presented with neurologic clinical signs had a severe lymphoplasmacytic meningoencephalitis with marked edema and variable necrosis. Remarkably, the lesions in the brain of the horses of this report resemble those described in horses infected by Trypanosoma brucei brucei, 12, 24, 28, 31 which causes a disease known as nagana, and those described for human trypanosomiasis caused by Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense. 9, 12, 23, 25, 27, 29, 35– 37
T. evansi antigen was detected by immunohistochemistry in the brain of 8 of 9 horses in the present report. Failure to detect antigen in 1 horse might have been caused by prolonged formalin fixation or autolysis. This immunohistochemistry technique had been used successfully for identification of T. evansi within brain lesions of cattle, 47 hog deer, 46 and buffaloes. 45 In the brains of horses from this report, protozoa were detected within blood vessels, in perivascular spaces, and in the brain parenchyma. This suggests that trypanosomes invaded the brain parenchyma and were responsible for the lesions in these horses. Thrombosis was not observed, so the pathogenesis of cerebral necrosis was not elucidated. Similarly, the mechanism by which trypanosomes entered the neuroparenchyma was not determined.
The introduction of T. evansi into a naïve population could explain the development of fatal trypanosomiasis with severe encephalitis in these horses. The reason for the sudden appearance of this disease in southern Brazil is unknown, but it could have been introduced by transportation of infected horses or migration of capybaras from regions where trypanosomiasis is enzootic. Another factor that may have contributed to the development of severe encephalitis was the use of subtherapeutic doses of diminazene aceturate and other antitrypanosomal drugs in the affected horses. 38 Several studies have demonstrated that the use of subtherapeutic doses of diminazene aceturate may prolong survival of horses experimentally infected by T. brucei spp., but it is associated with subsequent invasion of the central nervous system by trypanosomes and production of necrotizing encephalitis. 14, 18, 19, 26, 28, 34, 39, 44 Diminazene aceturate clears trypanosomes from tissues except in the central nervous system, 17 because the drug does not cross the blood-brain barrier (BBB). 19, 31 Thus, trypanosomes in the central nervous system survive antitrypanosomal therapy, and with a change in their surface glycoproteins may lead to new parasitemias. 17
How trypanosomes penetrate the BBB is unknown, but several mechanisms have been proposed: 1) entrance through sites where the BBB is incomplete, such as sensory ganglia and circumventricular organs; 29 2) deposition of immune complexes in the choroid plexus, with resultant increase in vascular permeability; and 3) release of toxic substances by trypanosomes that cause opening of intercellular tight junctions in the ependymal lining of the ventricular system. 20 It is believed that invasion of the central nervous system by trypanosomes occurs where the BBB has been disrupted, either directly by the parasites or by release of chemical mediators, including cytokines and proteases. 13, 23, 26 An interaction between the trypanosomes and the host response to them could promote tissue damage and facilitate parasitic entry into the central nervous system. 23 This scenario is supported by the histopathologic changes and immunohistochemistry in the present report.
Trypanosomiasis due to T. evansi should be considered in the differential diagnosis of encephalitis in horses in regions where the disease is enzootic. The differential diagnoses include equine herpesvirus type 1 myeloencephalopathy, Eastern equine encephalitis, Western equine encephalitis, Venezuelan equine encephalitis, equine protozoal myeloencephalitis, West Nile virus infection, and rabies. The nature and distribution of lesions in the central nervous system of horses with naturally occurring T. evansi infection should help to distinguish trypanosomiasis from viral or other protozoal infections.
Footnotes
Acknowledgements
This study was supported by the Brazilian Government (CNPq) grant No. 478779/2007-0. We are indebted to the following persons and institutions: Dr. Marta Teixeira, Universidade de Sao Paulo, Departamento de Parasitologia of the Instituto de Ciências Biomédicas, for the identification of Trypanosoma evansi; Dr. Rosangela Zacarias, Universidade Estadual Paulista, Departamento de Patologia Veterinária, for the performance and interpretation of enzyme-linked immunosorbent assay tests; The Prairie Diagnostic Service, University of Saskatchewan, Western College of Veterinary Medicine, for performing the immunohistochemistry; Dr. John Roths, Texas A&M University, Department of Veterinary Pathobiology, for imaging technical support; and Dr. Corrie Brown, University of Georgia, Department of Veterinary Pathology, for proofreading the manuscript.
Portions of this study were reported in Pesq Vet Bras