
Abstract
Congenital cystic adenomatoid malformation (CCAM) is a developmental lung abnormality characterized by abnormal proliferation of mesenchymal elements and failure of bronchiolar structures to mature, ultimately resulting in the compression of normal pulmonary tissue and mediastinal shift with rapid expansion of cysts. Although various clinical and pathologic studies of CCAM in humans exist, CCAM has yet to be reported in animals, even in nonhuman primates. In the present study, histopathologic analyses of a neonatal cynomolgus monkey that died 17 days after birth revealed that normal lung architecture was replaced by disorganized overgrowths of cysts lined with simple cuboidal epithelium. The epithelium projected a few ciliates into the air spaces and produced mucus. To our knowledge, this is the first case study describing CCAM or a CCAM-like lesion in nonhuman primates.
Congenital cystic adenomatoid malformation (CCAM) is a rare fetal developmental abnormality of the lung. First described by Ch'in and Tang in 1949, 2 it is characterized by abnormal development of terminal respiratory structures, resulting in an adenomatoid proliferation of bronchiolar elements and cyst formation. 11, 18, 19 CCAM is observed mainly during the neonatal period and infancy and can be diagnosed prenatally by ultrasonography after 16 weeks of gestation. 9, 10, 13, 14 CCAM lesions are believed to be a consequence of abnormal embryogenesis during the first 6 to 7 weeks of pregnacy. 18
Stoker et al. 18, 19 classified CCAM into three types based on clinical and histologic criteria. In the neonatal period, the expansion of cysts and the compression of surrounding lung parenchyma cause progressive respiratory distress. 18, 19 Moreover, emphysema-related lung masses also cause dyspnea, eventually leading to death. 18, 19 Fetuses with CCAM tend to be born prematurely or are stillborn. 12 Due to the severity and impact of this disorder, numerous clinical and pathologic studies in humans have focused on determining the pathologic mechanisms underlying CCAM and the clinical management of CCAM. 1, 4, 8, 15– 17 Although CCAM is well characterized in humans, CCAM or CCAM-like pathogenesis has not been reported in animals. To our knowledge, we report here the first case of CCAM in a nonhuman primate, as demonstrated through histopathologic analyses of a neonatal cynomolgus monkey.
A male cynomolgus monkey (Macaca fascicularis) born in our colony died naturally 17 days after birth. The animal was housed in an individual cage with its mother and maintained according to the National Institute of Biomedical Innovation rules and guidelines for experimental animal welfare. The mother monkey had the experience to raise another 6 baby monkeys. In this case, the mother monkey embraced her baby as usual, and the lactation also was observed. After birth, breeding staff members observed the baby monkey to check its health status; however, they could not find any abnormalities until 3 days before it died. Necropsy was performed. Tissues were fixed in 10% neutral-buffered formalin, processed conventionally, embedded in paraffin, cut into 3-μm–thick sections, and stained with hematoxylin and eosin. Lung tissues were also stained with periodic acid–Schiff (PAS), Alcian blue (AB), Masson's trichrome, and elastica van Gieson stains.
For immunohistochemical analyses, sections were first deparaffinized by pretreatment with 0.3% H2O2 solution. Next, sections underwent antigen retrieval with citric acid buffer and heating in an autoclave at 121°C for 10 minutes. Finally, sections were incubated free floating in primary antibody solution overnight at 4°C. The primary antibodies were anti-cytokeratin antibody (AE1/AE3 clone; 1:50; Dako, Glostrup, Denmark), anti-vimentin antibody (1:100; Dako), and anti–von Willebrand factor VIII antibody (1:400; Dako). Following brief washes with buffer, the sections were sequentially incubated with Polymer immunocomplex (Dako) for 30 minutes. Immunoreactive elements were visualized by treating the sections with 3-3′ diaminobenzidine tetroxide (Dojin Kagaku, Kumamoto, Japan). The sections then were counterstained with hematoxylin.
Macroscopically, lesions were confined to the right lower pulmonary lobe. In the lesion area, we found a thin-walled multicystic structure (3.5 cm × 2 cm × 1 cm) filled with air (Fig. 1). The other pulmonary lobes were pinkish-red and showed signs of atelectasis (Fig. 1). In heart, both atria dilated with clot. Microscopically, the affected lung displayed diffuse, abnormal architecture composed of numerous large, partially collapsed air spaces. These lesions looked like adenomatoids, and air spaces were lined with simple cuboidal epithelium surrounded by connective tissue and immature cartilage (Fig. 2). Within the air spaces, we found a few ciliates (Fig. 3), and PAS stained a little mucus on a part of epithelium (Fig. 4). AB weakly stained premature cartilage. Masson trichrome stained fibrous interstices, but smooth muscles were not observed. Elastica van Gieson stained elastic fibers around the air spaces. Other lung tissues were congested and were compressed by the surrounding mass. The border between the cystic lesion and normal tissue was not clear. In liver and kidney, congestion was observed.
Immunohistochemical analyses revealed that the epithelium of the lesion was immunopositive for cytokeratin but negative for vimentin and von Willebrand factor VIII. However, the fibrous interstices and premature cartilage of the lesion were immunopositive for vimentin.
The congenital pulmonary abnormalities can be divided into some classifications. 5, 6
1. Bronchopulmonary sequestration: 5, 6 bronchopulmonary sequestration is a pulmonary malformation in which a portion of lung parenchyma has no communication with the tracheobronchial tree and receives its blood supply via a systemic artery. The abnormality is observed as intralobar or extralobar. The first type (intralobar sequestration [ILS]) is contiguous with normal lung parenchyma and within the same visceral pleural envelope. The abnormal tissue is usually well demarcated from surrounding lung parenchyma and consists of one or more cystic spaces filled with mucus or pus. Microscopically, they resemble dilated bronchi, with respiratory epithelium and occasional mural cartilage plates. The latter type (extralobar sequestration [ELS]) is enclosed within its own pleural membrane, usually close to a normal lung. In addition to these types, the bronchopulmonary foregut malformation (BPFM) 7 is characterized by the pulmonary sequestrations that communicate with the upper gastrointestinal tract.
2. Bronchogenic cysts: 5, 6 Bronchogenic cysts are lesions of congenital origin derived from the primitive foregut and are usually solitary, thin-walled, unilocular, and roughly spherical. They are filled with either mucus or serous fluid, and they do not communicate with the trancheobronchial tree. Microscopically, the cyst wall is lined by respiratory epithelium and contains cartilage, smooth muscle and, sometimes, seromucinous bronchial-type glands.
3. Congenital lobar emphysema (CLE): 5, 6 CLE is characterized by severe overinflation of a pulmonary lobe. The pathogenesis is perhaps due to hypoplasia of bronchial cartilage. Microscopically, there is massive distention of alveolar spaces, but not tissue destruction.
4. Congenital cystic adenomatoid malformation (CCAM): 3, 5, 6, 18, 19 CCAM consists of an intralobar mass of disorganized pulmonary tissue that can exist with or without gross cyst formation. The anomaly is considered by some to represent a hamartoma, whereas others feel that it results from localized arrested development of the fetal bronchial tree. The cysts can usually be shown to communicate with normal airways; vascularization is by way of the pulmonary circulation. The histologic appearances give rise to the name, as there are numerous spaces of varying size, some of which resemble glandular acini, being lined cuboidal or columnar epithelium. According to the Stocker et al. 18, 19 on the basis of 38 cases, CCAM is classified into three categories. Type I consists of some large cysts (>2 cm in diameter). The lesions are lined by ciliated cuboidal or columnar epithelium with elastic tissue or smooth muscle. Mucus-producing cells or cartilage may be present. Relatively normal alveoli may be seen between or adjacent to these cysts. Type II consists of small cysts (<2 cm in diameter) lacking mucus-producing cells and cartilage. Type III consists of bulky, firm, solid mass with small cysts (<0.5 cm in diameter). The lesions are lined with nonciliated cuboidal epithelium.
In this case, we found the abnormal architecture composed of numerous large air spaces. These cysts looked like adenomatoids, and they were lined with simple cuboidal epithelium surrounded by elastic tissue and premature cartilage. These pathologic features are different from that of ILS, ELS, BPFM, bronchogenic cysts, or CLE, because cysts were not unilocular, they did not include mucus or serous fluid, and they had neither abnormal artery nor communication with the upper gastrointestinal tract. Furthermore, the lesions were not massive distentions of alveolar spaces. Taken together, this monkey case is considered CCAM.
We applied the Stocker et al. 18, 19 classification scheme to our monkey case, since no reports on CCAM in animals currently exist. In this monkey case, we observed large cysts greater than 2 cm in diameter (Fig. 1) lined with sparsely ciliated epithelium (Fig. 3) that produced a little mucus in a part (Fig. 4). These cysts also contained immature cartilage (Fig. 2). Thus, this monkey case exhibited sufficient pathologic features to meet the criteria for type I CCAM. We considered that pulmonary lobes were compressed by cysts and alveolars were collapsed, so that this monkey would suffer from respiratory distress and die. Moreover, the heart or great vessel may be compressed by pulmonary cysts, so that we consider liver and kidney congestion as a result of circulatory failure.
To our knowledge, this is the first case study describing CCAM or CCAM-like lesions in nonhuman primates. We have no information about the genetic background of the monkey in this case. Nonetheless, this case is quite noteworthy, because it provides an example of CCAM-like pathology in animal tissue. Should we encounter other CCAM-resembling or suspicious cases, we will carry out not only histopathologic analyses but also genetic analyses to determine the genetic background of these monkeys. Accumulating data from such analyses in nonhuman primates will contribute greatly to understanding CCAM pathology.
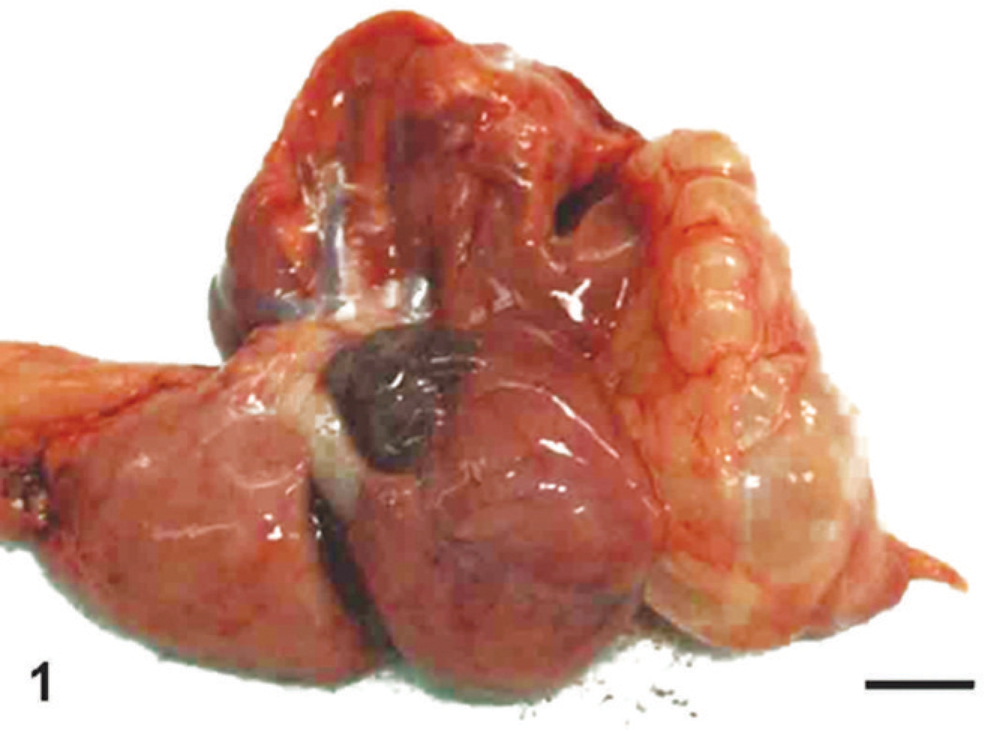
Lung, 17-day-old monkey. The right lower lobe contains a 3.5 cm × 2 cm × 1 cm, thin-walled, multicystic structure filled with air. The remaining lobes were pinkish-red and showed signs of atelectasis. Bar = 5 mm.
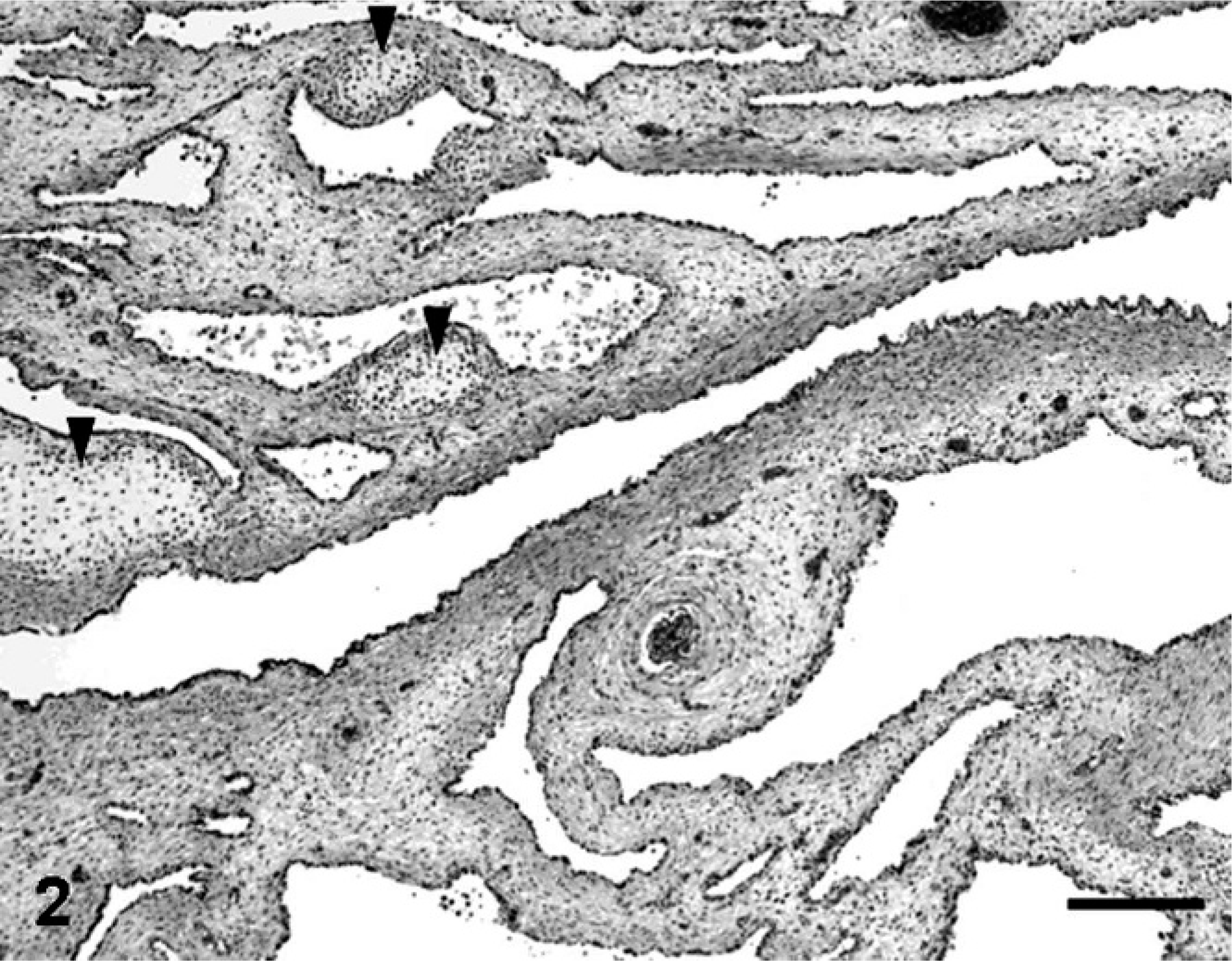
Lung, 17-day-old monkey. Photomicrograph of affected lung showing diffusely abnormal architecture characterized by numerous large air spaces lined with epithelium surrounded by connective tissue and immature cartilage (arrowheads). HE. Bar = 250 μm.
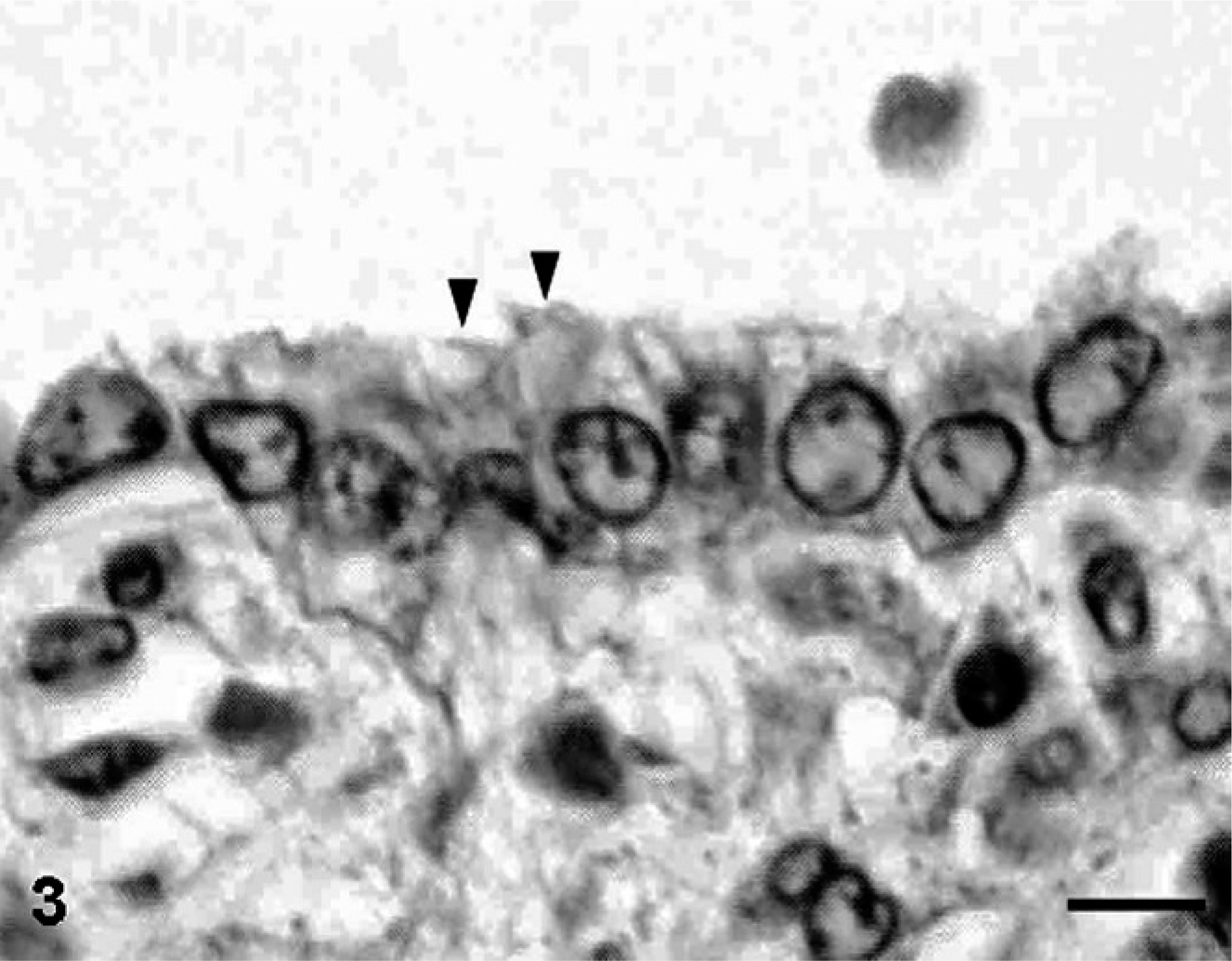
Lung, 17-day-old monkey. Air spaces are lined with simple cuboidal epithelium. A few ciliated cells (arrowheads) were observed within some air spaces. HE. Bar = 6 μm.
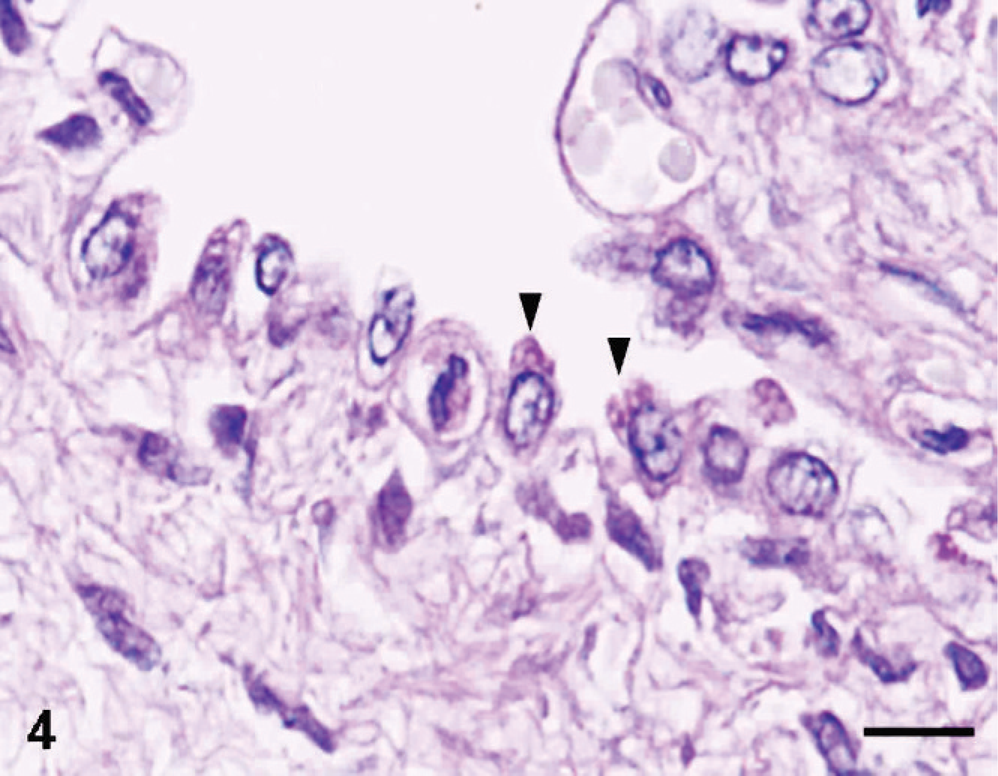
Lung, 17-day-old monkey. Periodic acid–Shiff demonstrated mucus (arrowheads) on a part of the epithelium lining air spaces. Bar = 8 μm.
Footnotes
Acknowledgements
This study was supported by Tsukuba Primate Research Center, National Institute of Biomedical Innovation, Japan.