
Abstract
Interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α) synergistically induce and sustain arthritis. Two competing hypotheses of arthritis induction are 1) that TNF preferentially mediates inflammation, whereas IL-1 impels bone destruction, or 2) that either cytokine controls the entire process. In this study, these propositions were tested in two experiments by instilling IL-1β or TNF-α into one knee of Lewis rats (n = 6/group) to incite arthritis, after which semiquantitative scores for inflammation, bone resorption, osteoclasts, and cartilage integrity were acquired. In the induction study, IL-1β or TNF-α (3, 10, or 30 μg) was given once to incite arthritis. After 2 days, IL-1β induced significant, dose-dependent increases in inflammation (mild to marked), bone resorption (minimal to moderate), and osteoclasts (minimal to moderate). In contrast, TNF-α induced minimal to mild inflammation but had little impact on resorption or osteoclasts. Both IL-1 and TNF (≥10 μg) yielded mild cartilage degeneration. Most lesion scores in TNF-treated rats were significantly lower than those in animals given the same dose of IL-1β. In the persistence study, rats were injected once with IL-1 or TNF (10 μg) and maintained for 2, 3, or 7 days. IL-1β significantly enhanced inflammation (all 3 days), bone resorption (days 2 and 3), osteoclasts (days 2 and 3), and cartilage matrix loss (days 2 and 3), whereas TNF-α augmented inflammation (days 2 and 3) and cartilage degeneration (day 2) but not bone resorption or osteoclasts. Thus, both IL-1β and TNF-α can launch inflammation, but IL-1β drives skeletal destruction.
Arthritis is initiated and sustained by the release of myriad proinflammatory cytokines (reviewed by Arend and Dayer 1 and Taylor 37 ). In patients with rheumatoid arthritis, the dominant proinflammatory cytokines are interleukin-1 (IL-1) (chiefly the inducible β form) and tumor necrosis factor-α (TNF-α). 2,7 Inhibition of IL-1β or TNF-α (or both) reduces the extent of inflammation in rheumatoid arthritis 9,33 and lessens inflammation and bone destruction in various experimental models of arthritis. 15,21,23,44,45 Therefore, biologic agents that specifically inhibit the action of these two cytokines are gaining rapid acceptance as early, aggressive treatments for rheumatoid arthritis. 6,10
IL-1β and TNF-α act synergistically to induce and maintain inflammation and skeletal erosion in arthritis. 8 Given the extensive coexpression of these two cytokines in joints with both acute 27 and advanced disease, 14,22,25 the precise role that each molecule plays in directing the early pathogenesis of arthritis remains unclear. Two competing propositions are that TNF-α preferentially drives acute inflammation, whereas IL-1β impels later bone destruction, 20,21,25 or that either IL-1β or TNF-α alone can control both inflammation and skeletal destruction. 15 The present experiments explored these competing hypotheses using direct intra-articular injection of purified cytokines. We selected this strategy so that the acute effects of each molecule could be studied in relative isolation from those related to the plethora of proinflammatory cytokines that are produced during the induction and progression of arthritis.
Materials and Methods
Experimental design
The current studies were designed to examine the respective functions of IL-1β and TNF-α in controlling the induction and maintenance of acute joint inflammation. These two cytokines were selected for study because they profoundly influence the course of rheumatoid arthritis, 2,7 where they participate both directly and indirectly (through induction of other proinflammatory molecules) in sustaining the disease. Two experiments were completed in series (Table 1): 1) an induction study assessing the impact of acute exposure to either IL-1β or TNF-α; and 2) a persistence study to define the time course during which intra- and periarticular lesions regress after a limited acute exposure to either IL-1β or TNF-α.
Experimental design for cytokine instillation studies.

Both cytokines were given once, as described below. The soluble mature forms of circulating IL-1β and TNF-α are both 17 kd, 32 so the two cytokines were given on an equimolar basis. The range of doses was extrapolated from previously reported data 12,18 to ensure a rapid and florid arthritic reaction.
These experiments were conducted in accordance with federal animal care guidelines and were preapproved by the Amgen Institutional Animal Care and Use Committee.
Animals and husbandry
Young, adult male Lewis rats (6–7 weeks and 175–200 g; Charles River, Wilmington, MA) were randomly assigned to treatment groups (n = 6/cohort) and housed two per filter-capped cage (room temperature 23 ± 2 C, relative humidity 50 ± 20%, 12-hour light-dark cycle). This strain was selected because of its demonstrated utility in arthritis investigations. Animals were given tap water and pelleted chow (#8640, Harlan Teklad, Madison, WI) ad libitum.
Cytokine instillation
Each rat was anesthetized with isoflurane (3%) in oxygen (4 liter/minute). Using a 28-gauge needle, the left knee was injected once through the infrapatellar ligament with 50 µl of filter-sterilized phosphate-buffered saline (PBS) containing either IL-1β or TNF-α. In a pilot study, we confirmed that the instillate was confined to the joint space by injecting Evan's blue dye (unpublished).
The recombinant human proteins (Amgen Inc., Thousand Oaks, CA) were expressed in Escherichia coli and purified by standard methods. Recombinant human versions were used for these experiments because of the high degree of homology between the human and rodent proteins 32 and the proinflammatory actions of either human IL-1β 12,13,24,36,43 or TNF-α 24,35,39,43 toward rat cells in vitro and in vivo. Endotoxin contamination was held below 10 EU (20 ng) per milligram protein because the lipopolysaccharide component is a potent inducer of both IL-1 and TNF-α.
Induction experiment
Either IL-1β or TNF-α was instilled once at 3, 10, or 30 µg. Rats were maintained for 2 days to study the dose response for initiation of joint lesions that develop in acute arthritis. This time point was selected based on a previous study demonstrating that joint damage peaks 2 days after cytokine injection. 41 An untreated normal control group was used rather than a vehicle-treated cohort for the first study. This labor-saving choice was deemed reasonable because in our experience normal young rats almost never have joint lesions (unpublished).
Persistence experiment
IL-1β, TNF-α (10 µg), or vehicle was injected once. Rats were kept for 2, 3, or 7 days to evaluate the progression of arthritis after a limited insult. A dose of 10 µg was chosen because results of the dose-response study indicated that, when given alone, both IL-1β and TNF-α induced biologically significant lesions at this level. A single administration was elected to limit secondary production of other cytokines, thereby allowing the effects of IL-1β and TNF-α to be studied in relative isolation.
Tissue processing
At necropsy, injected knees were separated from the foot and thigh, fixed intact by immersion in 70% ethanol, and then decalcified in a 1:1 mixture of 8 N formic acid and 1 N sodium formate. Almost all knees naturally assumed an angle of between 135° and 150° because severing the leg muscles prevented postmortem contraction. Next, knees were trimmed in the frontal plane by progressively removing tissue (in layers of 2 to 4 mm) from the rostral margin until the joint cavity and menisci were visible (internal landmarks, as shown in Fig. 1); the proper orientation was maintained by holding the razor blade parallel to the tibia when cutting the knee. For the few knees (about 5%) that contracted during fixation, the fascia and tendon insertions on the caudal surface were cut just above and below the joint so that the knee could be flexed to a 140° angle for trimming. Specimens were embedded in paraffin (rostral face down in cassette) and sectioned serially at 5 µm. One section was stained with toluidine blue because it imparts a uniform, deep purple color to normal cartilage. The other was subjected to an automated, indirect immunoperoxidase technique to label the osteoclast marker cathepsin K (CK) and then counterstained with hematoxylin and eosin (HE). 5 The immunohistochemical method used sequential incubations in CAS Block (Zymed Laboratories, San Francisco, CA) for 10 minutes, PBS containing a proprietary rabbit monoclonal anti-human CK antibody (1 µg/ml; Amgen) for 60 minutes, PBS with biotinylated goat polyclonal anti-rabbit antibody (1:200; Vector, Burlingame, CA) for 25 minutes, and peroxidase blocking solution (DAKO Corporation, Carpenteria, CA) for 25 minutes. An avidin–biotin–complex bridge was developed for 25 minutes using standard reagents (ABC Elite kit; Vector) and visualized using diaminobenzidine (DAB+ Substrate/Chromagen System; DAKO) for 3 minutes.
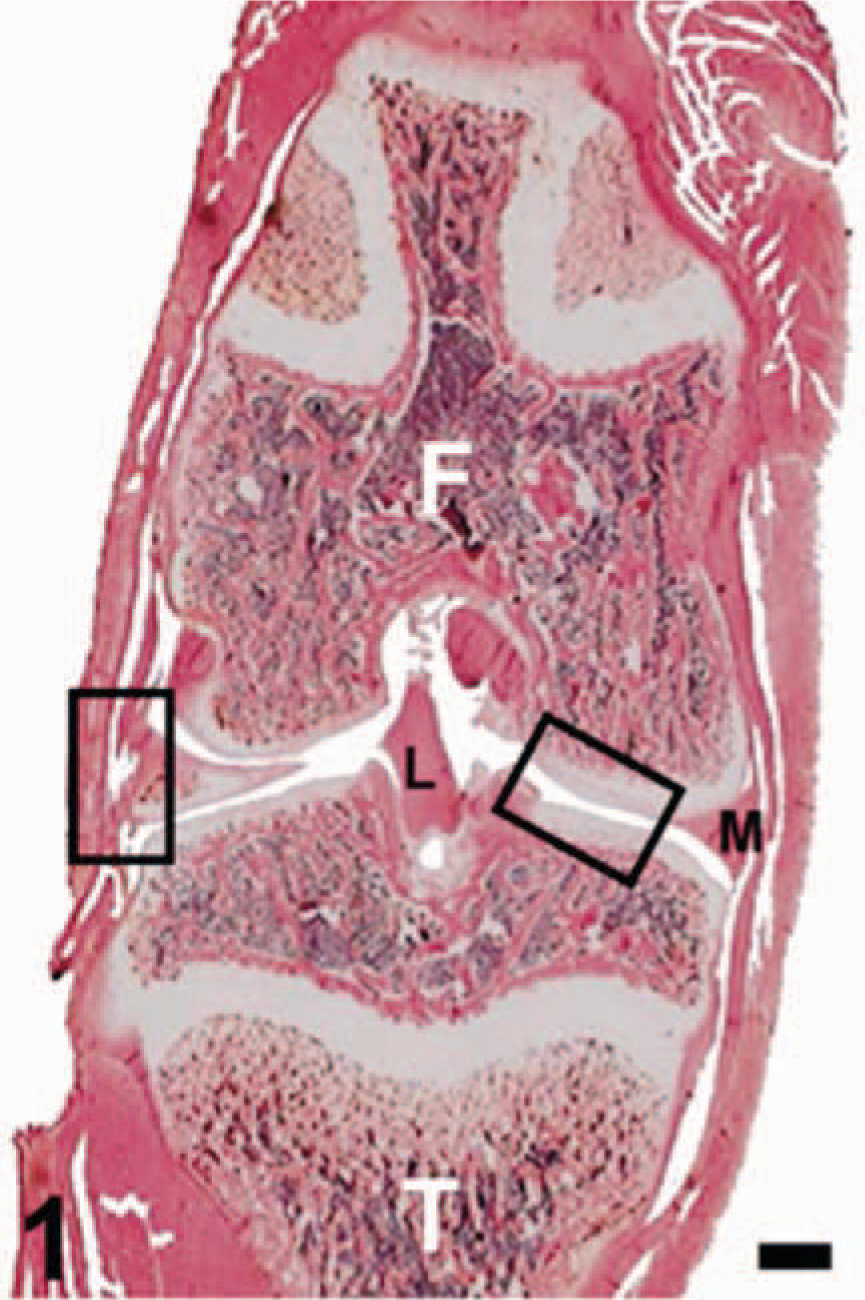
Knee (frontal section); normal rat. Abbreviations: F = femur; L = cruciate ligament; M = meniscus; T = tibia. Boxes denote regions shown at higher magnification in figures below. Anti-CK immunohistochemistry with HE counterstain. Bar = 1 mm.
Morphologic analysis
Four lesions were scored in each joint using tiered, semiquantitative criteria (Table 2). Cartilage degeneration was examined in toluidine-blue–stained sections. The other three changes—leukocyte infiltration, bone resorption, and subchondral osteoclasts—were evaluated in CK/HE–stained sections. These lesions were selected for analysis because such measures of inflammation, joint erosion, and cartilage matrix integrity commonly are assessed in animal models of arthritis. Separate scores for osteoclasts and bone resorption were acquired because we postulated that a rise in the population of bone-eroding cells was likely to precede actual bone damage in this acute arthritis model. Both sections for each animal were read consecutively (CK/HE followed by toluidine blue) so that data from each animal could be interpreted together. A “blinded” design was used to avoid bias because the three indices for scoring skeletal damage were based on subtle differentiations between grades.
Histopathology criteria for arthritis lesion scores.

Statistical analyses
The histopathologic lesion scores (ordinal variables) were analyzed by the chi-square test using commercially available software (JMP, v. 4.0; SAS Institute, Cary, NC). A P value of 0.05 was used to define significant differences between groups.
Results
Lesion character
The most prominent lesion induced by cytokine exposure was inflammation (Fig. 5). Two days after cytokine instillation, the reaction included neutrophils, with fewer lymphocytes and macrophages, and was most extensive in the periarticular soft tissues. Aggregates of neutrophils or fibrin (or both) were seen in some joints, particularly within recesses near the cruciate ligaments and beneath the patella. Over time, inflamed joints contained more mononuclear cells and less fibrin, but the neutrophil content remained high.

Knee; rat given IL-1β (10 µg) 2 days previously, demonstrating extensive inflammation in the periarticular soft tissues and joint cavity. Anti-CK with HE. Bar = 400 µm.
The primary skeletal changes seen in the inflamed joints were subchondral bone resorption accompanied by an enhanced subchondral population of activated osteoclasts (Fig. 6). Resorption was distinguished by more and deeper resorption (Howship's) lacunae lining the subchondral bone in both the femur and tibia. Osteoclasts filled most lacunae. The large osteoclasts (Fig. 6) in severely inflamed joints that had been injected with a cytokine typically had oval contours and multiple nuclei (an “activated” phenotype). 4 In contrast, osteoclasts from control knees (Fig. 3) or joints exhibiting a more modest inflammatory response (Fig. 9) generally had a fusiform or stellate conformation (a “resting” phenotype). 4 The appearance of the bone lesion and osteoclast population was similar at all time points after a single cytokine instillation.

Knee; normal rat. Frontal section of the joint cartilage and subchondral bone of the femoral (F) epiphysis (right [oblique] box, Fig. 1). Resting osteoclasts are rare, and resorption (Howship's) lacunae are shallow (arrowhead). Anti-CK with HE. Bar = 400 µm.
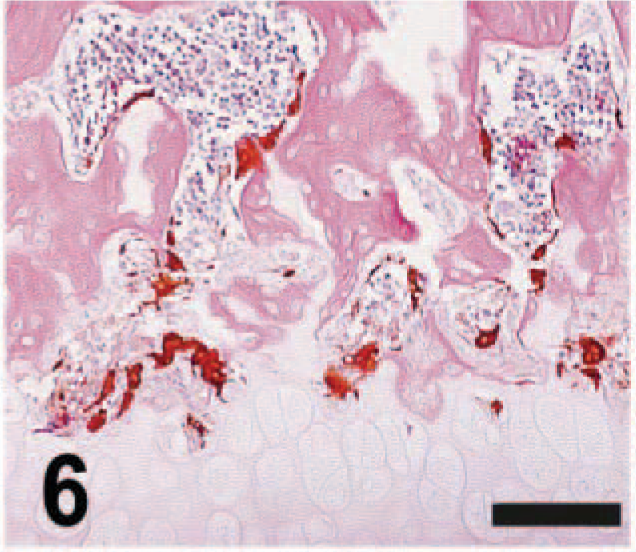
Knee; rat given IL-1β (10 µg) 2 days previously. The number and depth of resorption lacunae in the subchondral bone is increased, and the osteoclast population is both expanded and activated (as shown by their larger size and more rounded contours). Anti-CK with HE. Bar = 400 µm.

Knee; rat given TNF-α (10 µg) 2 days previously, showing increases in both resorption lacunae and osteoclasts. The osteoclast phenotype (fusiform to stellate conformation) suggests that these cells are differentiating but not yet fully activated (cf. Fig. 6). Anti-CK with HE. Bar = 400 µm.
Cartilage degeneration was induced in some joints after acute exposure to either cytokine. This change was exemplified by focal to multifocal loss of toluidine blue staining in the superficial cartilage matrix (Fig. 7); however, many cytokine-exposed joints remained unaffected (Fig. 10). The extent and severity of this lesion was higher in joints with more extensive inflammation—especially if the leukocyte infiltrate was present within the joint cavity. The degree of degeneration did not change with time after a single cytokine injection.

Knee; rat given IL-1β (30 µg) 2 days previously, revealing degeneration (loss of staining intensity [defined by arrowheads]) of the superficial matrix of the tibial joint cartilage. In this case, this change is associated with intra-articular accumulation of leukocytes (arrow). Toluidine blue. Bar = 150 µm.

Knee; rat given TNF-α (10 µg) 2 days previously. Note the extremely modest infiltration of leukocytes into the periarticular soft tissues relative to an equimolar IL-1β dose (Fig. 5). Anti-CK with HE. Bar = 400 µm.

Knee; rat given TNF-α (10 µg) 2 days previously, showing the essentially normal staining pattern observed in joints exposed acutely to lower doses of either cytokine. Toluidine blue. Bar = 150 µm.
Arthritis induction
Relative to uninjected joints (Figs. 2–4) led to substantial inflammation, subchondral bone resorption, and osteoclast expansion as well as some cartilage matrix degeneration 2 days after instillation. The rise in the lesion score severity was dose dependent for both cytokines.

Knee; normal rat. Frontal section magnifying the meniscus (M) and associated periarticular tissues (left [vertical] box, Fig. 1). Anti-CK with HE. Bar = 400 µm.

Knee; normal rat. Frontal section of opposing femoral (F) and tibial (T) joint cartilages (right [oblique] box, Fig. 1), showing uniform staining intensity of the cartilage matrix in all zones. The tip of the meniscus (M) routinely exhibits less intense labeling. Toluidine blue stain for cartilage matrix integrity. Bar = 150 µm.
All three IL-1β doses elicited significant increases in inflammation (Fig. 11), bone resorption (Fig. 12), and osteoclasts (Fig. 13). The increase in inflammation was mild to marked, whereas those for bone resorption and osteoclasts were minimal to moderate. The cartilage score was enhanced significantly to a minimal degree for IL-1β doses ≥10 µg (Fig. 14). In contrast, all three TNF-α doses induced significant but minimal to mild inflammation (Fig. 11) but did not affect bone resorption (Fig. 12) or osteoclasts (Fig. 13). The cartilage score was enhanced significantly to a minimal degree for TNF-α at 10 µg (Fig. 14). For a given dose, IL-1–treated rats had significantly greater scores for inflammation (doses ≥10 µg), bone resorption (all doses), and osteoclasts (all doses) 2 days after treatment than did animals injected with TNF.

Inflammation.

Bone resorption.

Osteoclast expansion.

Cartilage degeneration. Induction of acute arthritis by a single intra-articular instillation of IL-1β or TNF-α (3, 10, or 30 µg of purified protein) 2 days previously. Numbers inside the bars or just above the error bars indicate the group mean inflammation score. “1” denotes a significantly greater lesion (P ≤ 0.05) relative to untreated control rats and “2” a significant difference (P ≤ 0.05) between the changes induced by equimolar doses of IL-1β and TNF-α.
Arthritis persistence
IL-1β or TNF-α (10 µg) produced significant, sustained increases in scores for inflammation (both cytokines), bone resorption (IL-1 only), osteoclast expansion (IL-1 only), and cartilage matrix degeneration (both cytokines) 2, 3, or 7 days after instillation into the femorotibial joint cavity. Lesions induced by both cytokines regressed during the 7-day course of this experiment, indicating that arthritis induced by a limited cytokine exposure is self-limiting.
For IL-1β, the extent of inflammation was mild 2 and 3 days after injection and minimal after 7 days (Fig. 15). IL-1β also enhanced bone resorption (Fig. 16) and osteoclasts (Fig. 17) to a minimal to mild degree at 2 and 3 days after instillation but not after 7 days; minimal cartilage degeneration occurred in this same pattern (Fig. 18). In contrast, TNF-α induced minimal to mild inflammation only at the two early time points (Fig. 15). The only other lesion of significance elicited by TNF was minimal loss of cartilage matrix after 2 days (Fig. 18). A single TNF exposure did not affect bone resorption (Fig. 16) or osteoclasts (Fig. 17). IL-1β generated significantly greater inflammation, bone resorption, and osteoclasts than did TNF-α 3 days after cytokine treatment.

Inflammation.

Bone resorption.

Osteoclast expansion.

Cartilage degeneration. Persistance of acute arthritis by a single intra-articular instillation of IL-1β or TNF-α (10 µg of purified protein). Numbers inside the bars or just above the error bars indicate the group mean lesion score. “1” denotes a significantly greater lesion (P ≤ 0.05) relative to untreated control rats and “2” a significant difference (P ≤ 0.05) between the changes induced by equimolar doses of IL-1β and TNF-α. Abbreviation: d = days after injection.
Discussion
Inflammation and skeletal destruction in arthritis are determined by interactions between numerous pro- and anti-inflammatory molecules. On the proinflammatory side, two major players are IL-1β and TNF-α. Importantly, many studies indicate that IL-1β and TNF-α act cooperatively to promote joint inflammation. For example, IL-1 and TNF-α exhibit synergism in vitro when inducing production of plasminogen activator by human articular chondrocytes, 11 or collagenase and prostaglandin E2, 3,26 IL-6, 16 or IL-8 31 by human synovial fibroblasts. In vivo, IL-1 and TNF-α act synergistically to recruit leukocytes into rabbit joints. 18 In like manner, molecules that bind IL-1 or TNF-α—thereby blocking signal transduction through these two pathways—exhibit synergistic efficacy in decreasing the severity of inflammation and bone destruction in the Lewis rat model of adjuvant arthritis. 15 The synergism between IL-1β and TNF-α has been suggested to result from reciprocal positive feedback cycles by which IL-1 induces TNF-α 19 and TNF-α induces IL-1; 38 IL-1 has also been shown to induce itself. 34 Given the extensive colocalization of these two molecules in advanced disease, 14,22,25 the precise role that each molecule plays in the pathogenesis of acute arthritis in vivo remains unclear.
We undertook the present study to compare the effects elicited by IL-1β and TNF-α during the induction and early maintenance phases of acute arthritis. In particular, we sought evidence to select between two contending postulates: preferential control of inflammation by TNF-α and bone damage by IL-1β 21,42 or co-regulation of both inflammation and bone destruction by the actions of either IL-1 or TNF-α. 15,30 Our present data show that one exposure to equimolar doses of either IL-1β or TNF-α initiates acute arthritis, thus demonstrating that both cytokines can launch disease. Interestingly, however, our findings also show that IL-1β impels joint lesions to a substantially greater degree and for a longer time than does an equivalent dose of TNF-α. Taken together, these data provide support for the second hypothesis. Still, IL-1β was a more potent stimulus for subchondral bone resorption and osteoclast expansion than was TNF-α. Thus, in this respect, our data also confirm the first hypothesis because IL-1β clearly plays a greater role in the genesis of arthritis-associated skeletal destruction.
One potential criticism of the current experiments is that the cytokine levels used in this study were high relative to those needed to initiate a limited arthritic response. For example, IL-1 induces proteoglycan loss in cartilage matrix and leukocyte infiltration when injected into knees of mice or rabbits 17,28 at doses of 25 ng or less. In like manner, intra-articular injection of proarthritic materials yields local production of IL-1 and TNF-α at picogram levels. 29,40 However, we purposely selected the cytokine doses for the present study based on the relative insensitivity of the rat knee to the proinflammatory influence of IL-1 as arthritis reportedly is initiated in this joint only if the intra-articular dose of recombinant human IL-1 exceeds 90 µg. 12 An additional consideration was that administration of a high dose reasonably would be expected to overwhelm the effects of other proinflammatory molecules induced by IL-1β or TNF-α, thereby allowing the true proarthritic potential of each cytokine to be assessed. Our data show that the range of doses we chose to administer produced a modest, self-limiting arthritis, which shows that a single IL-1β or TNF-α exposure of ≤30 µg was not sufficient to induce the secondary cytokine cascades necessary to sustain the inflammatory process.
A potential explanation for the divergent proarthritic responses between IL-1 and TNF in the present study is that human IL-1β can stimulate rat-derived inflammatory cells, whereas human TNF-α cannot. This contention is supported in a general sense by data on rats showing that the stimulatory effect of human IL-1β toward the hypothalamic-pituitary-adrenal axis exceeds that engendered by human TNF-α. 43 The maximum cytokine dose used in the endocrine study (3 µg/rat) was 10-fold lower than the highest dose given during the present experiments (30 µg/rat); furthermore, the endocrine study used intravenous delivery, which would have greatly diluted the cytokine levels at the target tissue relative to our localized intra-articular injection. A review of recent literature amply demonstrates that human IL-1β and human TNF-α can enhance the activity of proinflammatory pathways in rat-derived cells and tissues, 13,36,39 which supports our interpretation that the discrepancy between the acute proarthritic responses of these two molecules in our rat intra-articular injection model represents a true biologic effect. However, with the exception of our present experiments, the functional consequences of exposing tissues from rat joints to recombinant human versions of IL-1β or TNF-α apparently have not been tested in a head-to-head experiment under either in vitro or in vivo conditions. Additional work with this and other arthritis models using various cytokine doses should provide considerable assistance in elucidating the specific in vivo functions of IL-1β versus TNF-α, as well as for comparing the effects of additional pro- and anti-inflammatory molecules in the onset and progression of acute arthritis.
Footnotes
1A part of this study was presented at a symposium on “Immune Mechanisms and Disease” (sponsored jointly by the New York Academy of Sciences and the Henry Kunkel Society) at St. Georges, Grenada, West Indies, on 14–17 April 2002 (extended abstract published in Ann NY Acad Sci 987:295–298, 2003).