
Abstract
Necropsy of two American Saddlebred fillies diagnosed with epitheliogenesis imperfecta (EI) revealed missing patches of epithelium of the skin and oral mucosa as well as dental abnormalities. Examination of the digestive tract did not reveal signs of pyloric atresia in either foal. Histopathologic examination revealed separation of the epidermis from the dermis. In both foals a division within the lamina lucida of the basal lamina was observed by transmission electron microscopy. In comparison with an age-specific control, the ultrastructure of intact skin from the EI-affected foals showed abnormal hemidesmosomes, which lacked a subbasal plate. The morphological and ultrastructural defects observed in the EI-affected American Saddlebred foals were similar to those observed in Herlitz junctional epidermolysis bullosa-affected human newborns, which is caused by a defect in one of the subunits of laminin-5. The close similarity of lesions of the human and equine diseases suggests that EI may be caused by a laminin-5 defect.
Equine epitheliogenesis imperfecta (EI) is a recessive hereditary mechanobullous neonatal disease characterized by missing epithelium on the skin and oral mucosa. These lesions vary in size and location but usually consist of irregular patches of missing hair and epithelium on the legs and back exposing the underlying dermis.12 Frequently, affected foals have patchy areas in the oral mucosa and tongue where the epithelium is absent. The teeth may also be affected with irregular and pitted lesions on the surface.6 Affected foals are ambulatory and usually display only a brief interest in feeding, most likely because of oral sensitivity. There is no known treatment for EI, and affected foals are usually euthanized soon after birth.
An analogous hereditary disease called epitheliogenesis imperfecta neonatorum bovum was first proposed as a distinct disease in cattle in 1928.8 Hadley described several cases of hereditary defects in the epithelium of newborn Holstein calves. He observed “defective formation of skin below the knee and hocks, defects in the integument of the muzzle and in the mucous membrane of the nostrils, tongue, hard palate, and cheeks.”8 The pathological entity, which came to be known as EI, was first described in horses of unknown breed in Germany in 1913.7 In 1935, a similar defect of the epithelium was described in Ardennes foals in Sweden.3 On the basis of a pedigree analysis and anecdotal evidence of the foaling of similarly affected foals from the same sire and dam, Berthelsen et al.3 proposed that EI had a genetic cause and a recessive pattern of inheritance. It was not until 1957 that Butz et al.5 described EI in cold blood horses in Germany and provided further evidence, through breeding experiments, of an autosomal recessive pattern of inheritance.
The first reported case of EI in American Saddlebred horses occurred in 1975 and was reported to the American Saddlebred Horse Association (ASHA). Currently 34 verifiable cases have been reported to the ASHA. Construction of a partial pedigree for American Saddlebreds showed a pattern of inheritance and a frequency of occurrence of EI that was consistent with an autosomal recessive inheritance pattern (Lieto, personal observation). A genotypic frequency for EI of 0.0016 was estimated for the American Saddlebred horse population on the basis of the average yearly number of American Saddlebred foals registered by the ASHA (∼3,000) divided by the highest total number of EI-affected foals reported to the University of Kentucky (5) in a single year. An allelic frequency of 0.04 was calculated from the genotypic frequency using the Hardy-Weinberg equilibrium law (Lieto, personal observation). This estimate suggests that approximately 4% of the American Saddlebred breeding population carries an allele for EI.
Filly Nos. 1 and 2 were American Saddlebred foals that were diagnosed at birth with EI lesions. The foals were euthanized and presented for postmortem evaluation to the Animal Pathology Laboratory, Department of Veterinary Science, University of Kentucky. Examination of the two foals revealed that the distribution of missing epithelium was consistent with a diagnosis of EI. The two foals had both extensive lesions along the back and hindquarters and minimal signs of inflammation. Filly No. 1 had extensive areas of missing skin below the knees and hocks on all four legs, whereas in Filly No. 2 both the front legs were almost completely devoid of skin below the knees and the rear legs had several small patches of missing skin. In both foals, lesions were observed in the oral mucosa of the tongue, cheeks, and hard palate, as well as in the anal and vaginal mucosa. In addition, the teeth were partially erupted and the enamel had a rough and pitted appearance. Examination of the esophagus, stomach, and intestinal tract did not reveal any abnormalities in the epithelium or signs of pyloric atresia. Tissue samples were collected from the brain, liver, spleen, kidney, thymus, lung, and skin and prepared for histologic examination following standard protocols.13 Histopathologic examination of the tissue specimens obtained from internal organs by light microscopy did not reveal any abnormalities. However, examination of the skin from affected areas revealed subepidermal splitting with an intact epidermis in the blister roof (Fig. 1). The separation left both the epidermis and dermis intact and was along the plane of the basal lamina. This split was similar to lesions observed in humans affected with hereditary epidermolysis bullosa (EB). Human EB is a form of aplasia cutis congenita characterized by the congenital absence of the skin. There are several subtypes of varying severity (simplex, junctional, dystrophic) that can be differentiated by the depth of the split within the basement membrane. Determination of the precise location of the epidermis/dermis separation by transmission electron microscopy examination allows the diagnosis of the specific subtype of EB. In simplex EB, tissue separation occurs within the epidermal basal keratinocytes adjacent to the basal lamina. Junctional EB blister formation arises within the lamina lucida of the basal lamina, leaving the lamina densa attached to the underlying dermis. Finally, in dystrophic EB, tissue cleavage occurs within or below the lamina densa, which leaves the lamina lucida affixed to the epidermis.
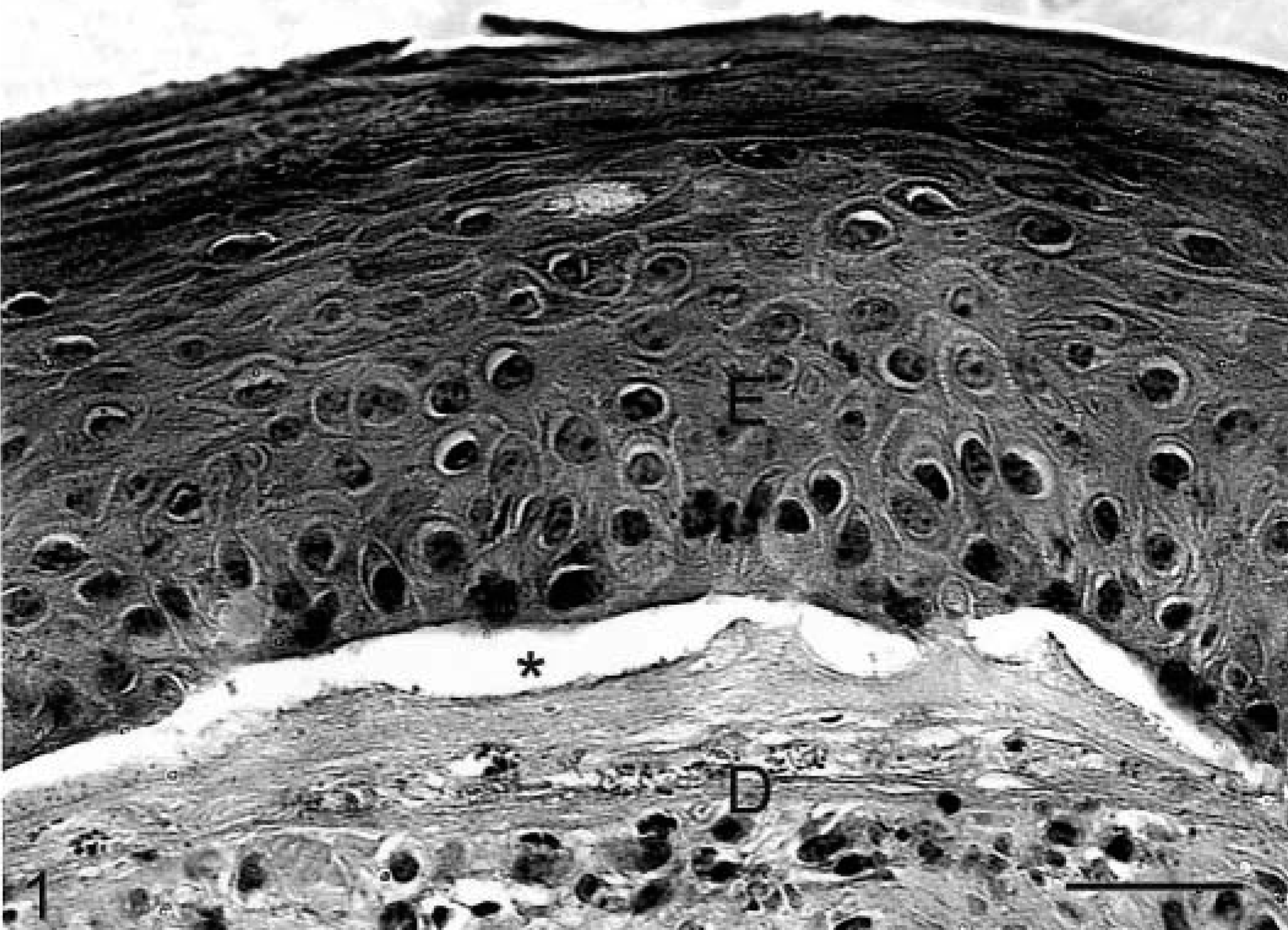
Skin; EI-affected foal No. 1. A ∗ indicates the location of a bulla within the basal lamina between the epidermis (E) and the dermis (D). HE counterstain. Bar = 50 μm.
Tissue specimens from surface lesions and uninterrupted epithelium were removed for ultrastructural examination within 2 hours of euthanasia. Control skin samples were obtained within 6 hours after death from the hindquarters of a normal control, a male Thoroughbred foal without skin lesions, which died from neonatal asphyxia shortly after birth and was submitted for postmortem examination to the Livestock Disease Diagnostic Center, University of Kentucky. These samples served as an age-specific, EI-unaffected control for comparison with the affected EI foal samples. All samples were fixed in glutaraldehyde, stained with osmium tetroxide, embedded in epon and araldite resin, sectioned, and stained with lead citrate and uranyl acetate.9
To avoid duplication, images from only EI-affected American Saddlebred foal No. 1 are shown (Figs. 2, 5, 6); however, both animals presented similar lesions. The epidermal basal lamina of the normal newborn Thoroughbred foal contained numerous distinct, fully formed hemidesmosomes (Fig. 3). The interface between the epidermis and dermis displays the characteristic interdigitations of normal skin. At higher magnifications, the hemidesmosomes have clearly defined boundaries and a distinct hemidesmosome subbasal dense plate within the lamina lucida (Fig. 4). In normal mammalian skin, collagen-VII fibrils lie below the lamina densa, which bind to and interact with the laminin-5–anchoring filaments. These filaments traverse the lamina lucida and interact with α6β4-integrin, a hemidesmosome constituent.10 α6β4-Integrin is a surface membrane protein found on basal keratinocytes.
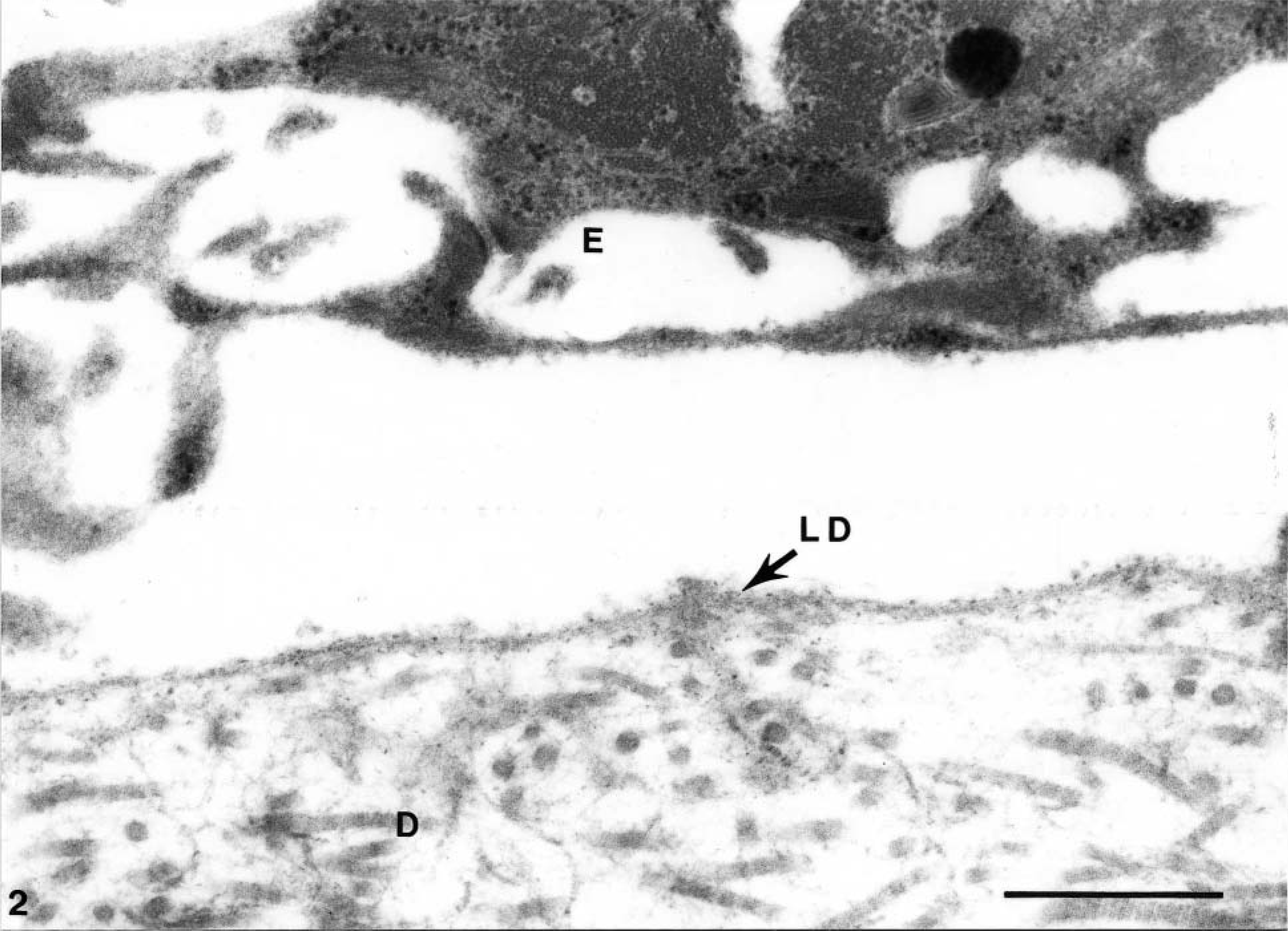
Electron micrograph. Skin basal lamina bulla; EI-affected foal No. 1. Higher magnification view of the bulla, similar to that displayed in Fig. 1, within the lamina lucida of the basal lamina, between the epidermis (E) and the dermis (D). The plasma membrane of the epidermal basal keratinocytes is intact and the lamina densa (LD) is attached to the dermis. Bar = 0.5 μm.
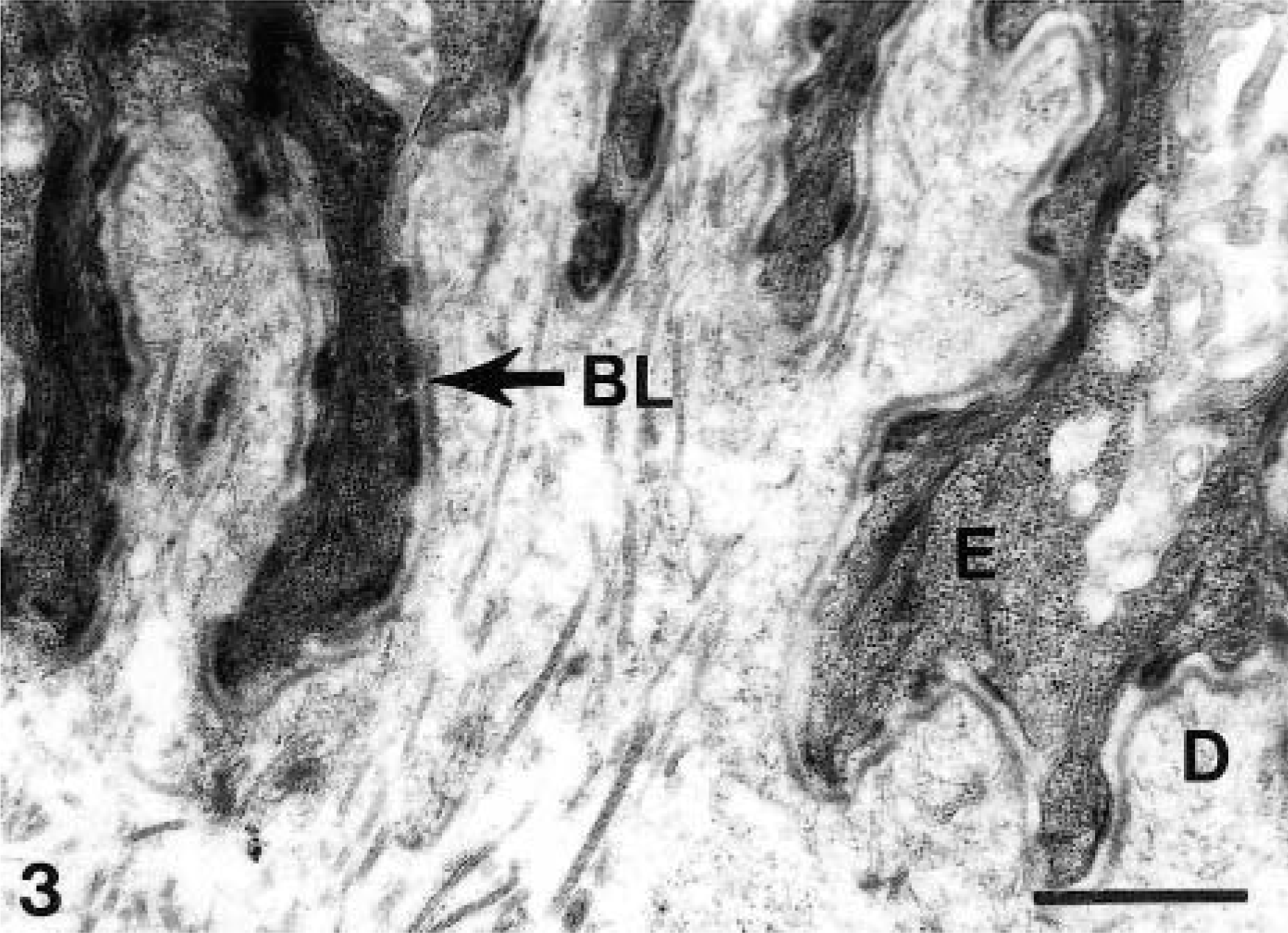
Electron micrograph. Intact skin; normal foal. In normal skin, complex interdigitations along the basal lamina (BL) between the epidermis (E) and the dermis (D) can be observed, as well as numerous, distinct, prominent hemidesmosomes. Bar = 1 μm.
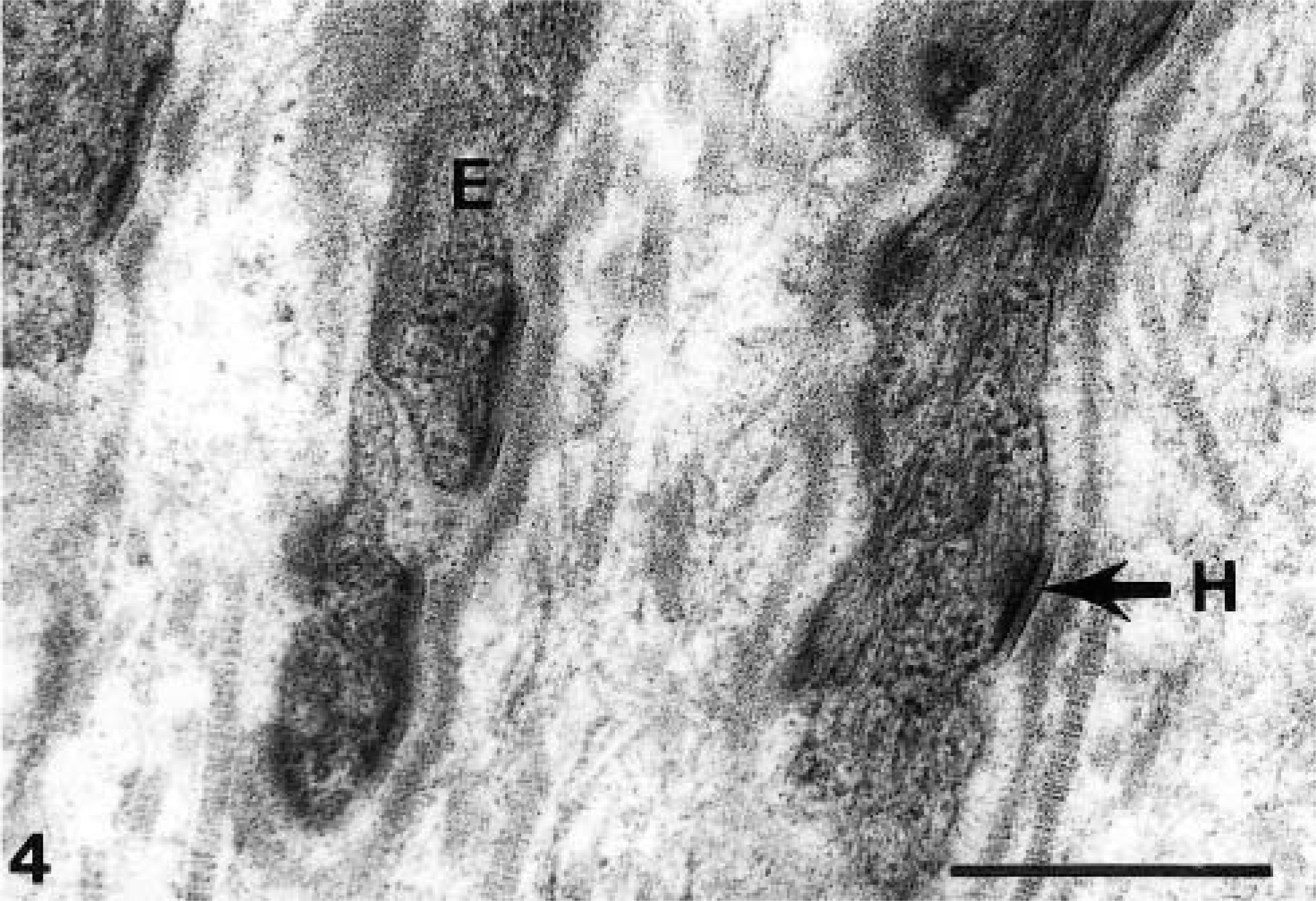
Electron micrograph. Intact skin; normal foal. Note the appearance of the hemidesmosomes at higher magnification, especially the subbasal dense plate and the attachment filaments of the indicated hemidesmosome (H). Bar = 0.5 μm.
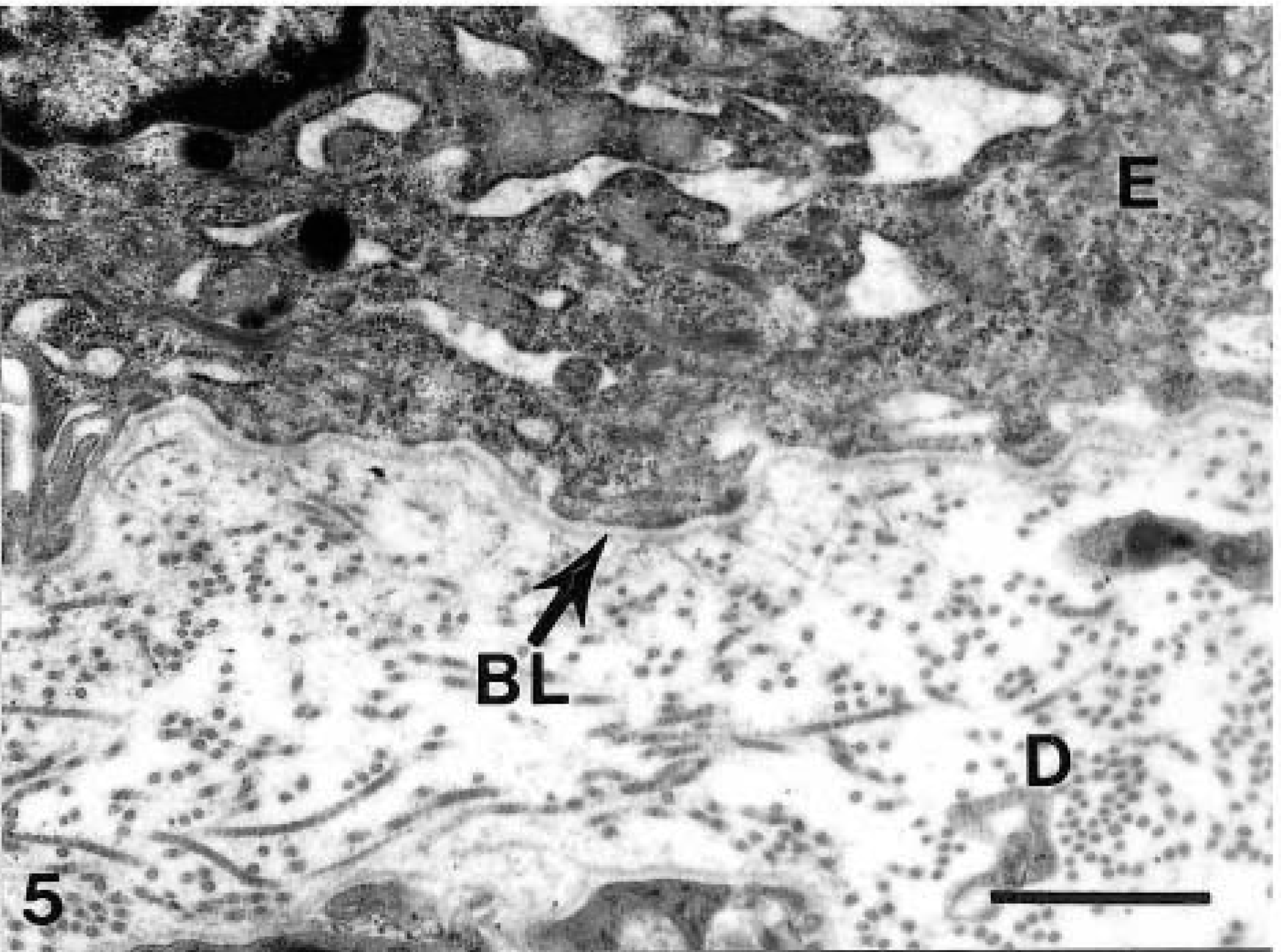
Electron micrograph. Intact skin section; EI-affected foal No. 1. Note the few small hemidesmosomes and their less prominent appearance in comparison with those in Fig. 3. Bar = 1 μm.
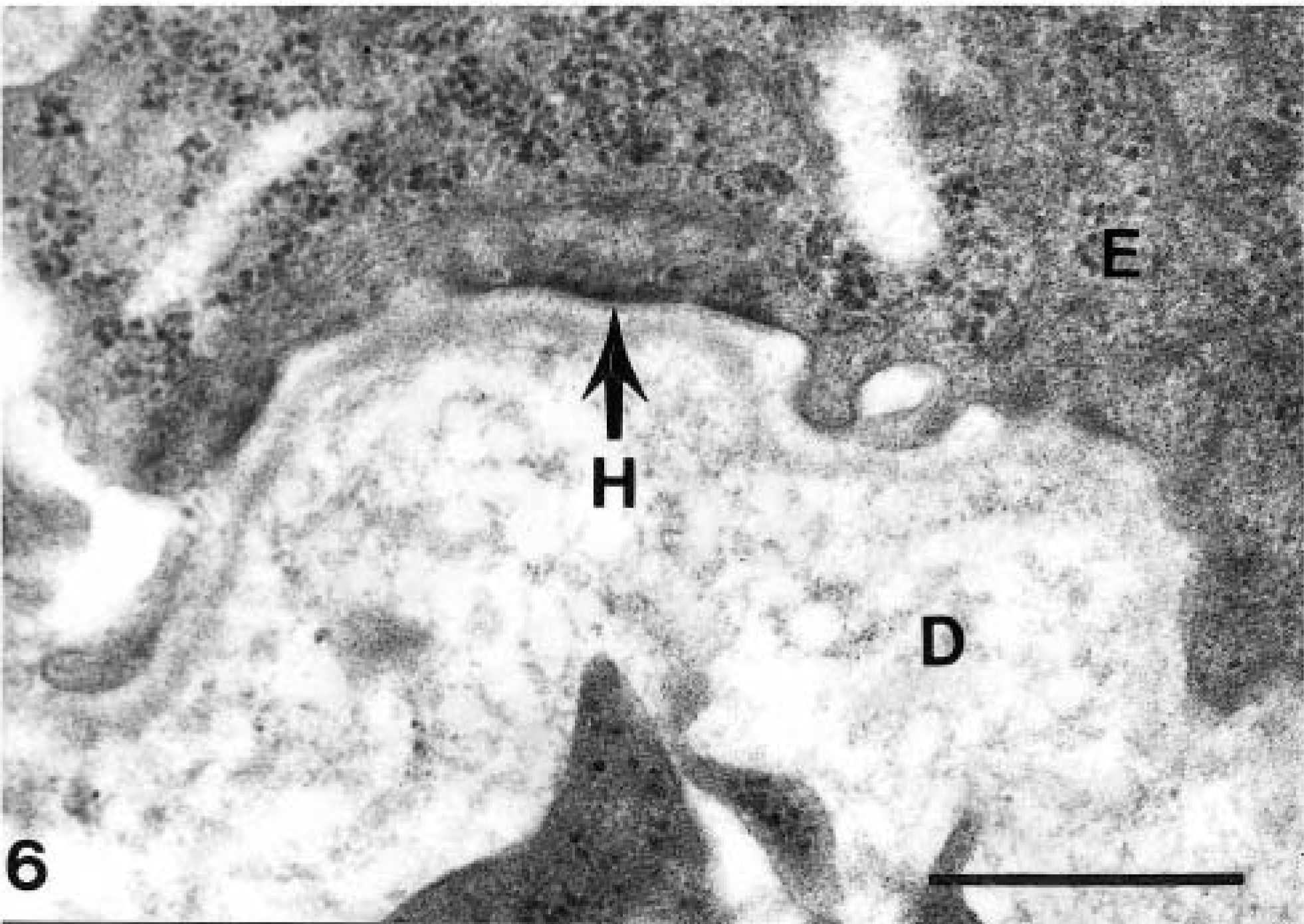
Electron micrograph. Intact skin section; EI-affected foal No. 1. Note the lack of a subbasal dense plate in the indicated hemidesmosome (H) as well as the small size and disorganized appearance of the hemidesmosomes. Bar = 0.5 μm.
The intact basal lamina of the two EI-affected saddlebred foals presents a starkly different view of hemidesmosome structure. The boundary between the epidermis and dermis in the attached lesion-free skin of the two affected foals had less complex interdigitation than that of the normal foal (Fig. 5). There were fewer hemidesmosomes, which appeared smaller in size and had poorly defined structure. The borders of the hemidesmosome were not clearly demarcated, and the subbasal dense plate was absent (Fig. 6). The epidermis was separated from the dermis within the lamina lucida of the basal lamina at the boundaries of skin lesions (Fig. 2). The lamina densa remained attached to the dermis, and the plasma membranes of the basal keratinocytes were intact (Fig. 2).
The observed separation within the lamina lucida and the overall skin morphology of both the EI-affected American Saddlebred foals are quite similar to a recessive hereditary disease in humans known as Herlitz junctional epidermolysis bullosa (HJEB). In humans, this variant of junctional epidermolysis bullosa is characterized by widespread epithelial lesions of the skin and oral mucosa in newborns and by increased mortality.2 These lesions are generally located on the back, buttocks, and bilaterally on the lower legs and upper arms. Human neonates with HJEB also have enamel defects with severe pitting.15 The distribution of surface skin lesions observed at birth in HJEB-affected human neonates is similar in pattern to those observed in the EI-affected American Saddlebred foals. For example, lesions observed on one extremity generally also occur on the opposite extremity in both American Saddlebred horses and in humans. These lesions probably form in utero as a result of chafing and damage to the epithelium. HJEB-affected neonates have a distinct separation of the epidermis from the dermis within the lamina lucida of the basal lamina. The intact skin of affected newborns contains abnormal hemidesmosomes that lack subbasal dense plates.4,11 HJEB is caused by a premature stop codon in one of the subunits of the laminin-5 heterotrimer (laminin-α3, laminin-β3, laminin-γ2) that results in a lack of expression of the respective protein chain.1,2,14 The laminin-5 heterotrimer forms the anchoring filaments that attach the α6β4-integrin of the hemidesmosome to the collagen-VII–anchoring fibrils. Mutations within either subunit of the α6β4-integrin can lead to a moderately severe form of junctional epidermolysis bullosa in humans that is usually accompanied by pyloric atresia. Necropsy evaluation of the two EI-affected American Saddlebred foals revealed no intestinal abnormalities. This observation, coupled with the general severity of the skin lesions in the two foals, suggests that EI in American Saddlebreds is not caused by a defect in the α6β4-integrin.
Hereditary dystrophic EB, in humans, is caused by mutations within the collagen-VII gene that lead to defective anchoring fibrils. On account of these defective anchoring fibrils the subepidermal split occurs within or below the lamina densa, leaving the lamina lucida attached to the roof of the epidermis.
The close similarity in the morphology and histopathology of EI-affected American Saddlebreds foals and HJEB-affected newborns is consistent with the possibility that equine EI in American Saddlebred horses may be caused by a laminin-5 defect. Further work is required to definitively determine if a mutation in one of the laminin-5 subunits causes EI in American Saddlebred horses. However, the evidence suggests that equine EI in American Saddlebreds is a lethal variant of junctional epidermolysis bullosa.
Footnotes
Acknowledgements
The authors would like to thank Mary Gail Engle and Pat Van Meter for their assistance with the transmission electron microscope imagery. This project was funded in part by The United Professional Horsemen's Association, The American Saddlebred Horse Association, and The Belgian Draft Horse Corporation. This study is part of a project of the University of Kentucky Agriculture Experimental Station and is published as paper number 02-14-62.