
Abstract
A canine model of glycogen storage disease Ia (GSD Ia), similar clinically, biochemically, and pathologically to the human disease, was established by crossbreeding Maltese and Beagle dogs carrying a mutated, defective glucose-6-phosphatase (G-6-Pase) gene. Ten puppies were born in three litters from these crossbreedings. Six were homozygous for the previously described M121I GSD Ia mutation. Of these six affecteds, two were stillborn, and one died at 2, 32, and 60 days of life, respectively (puppies A, B, C, D, E), while one is alive at age 15 months (puppy F). Affected puppies exhibited tremors, weakness, and neurologic signs when hypoglycemic. They had postnatal growth retardation and progressive hepatomegaly. Biochemical abnormalities included fasting hypoglycemia, hyperlactacidemia, hypercholesterolemia, hypertriglyceridemia, and hyperuricemia. Microscopic examination of tissues from affected puppies showed diffuse, marked hepatocellular vacuolation, with distended clear hepatocytes and central to marginally located rounded nuclei. In the kidneys of puppies D and E, there was segmental glomerular sclerosis and vacuolation of proximal convoluted tubular epithelium. Biochemical analysis revealed increased liver glycogen content and isolated markedly reduced G-6-Pase enzyme activity in liver and kidney. The canine G-6-Pase gene was characterized by screening a canine genomic library. It spans ~ 11.8 kb and consists of five exons with >90% amino acid sequence homology to the derived human sequence. The first 1.5 kb of the 5′ region was sequenced and contains several putative response element motifs homologous to the human 5′ region. Establishment of this canine colony of GSD Ia that closely resembles human disease and isolation of the canine genomic gene provides an excellent model for studying pathophysiology and long-term complications and an opportunity to develop novel therapeutic approaches such as drug and gene therapy.
Keywords
Glucose-6-phosphatase (G-6-Pase) is a key enzyme in glucose homeostasis catalyzing the terminal steps in both gluconeogenesis and glycogenolysis. The enzyme is present in high levels in liver, kidney, and intestine. G-6-Pase is a multicomponent system comprising a catalytic subunit located in the lumen of the endoplasmic reticulum and, presumably, other transport proteins that allow the passage of substrates and products across the endoplasmic reticulum membrane. The catalytic subunit is highly hydrophobic, tightly bound to the endoplasmic reticulum, and has a molecular mass of 36.5 kDa. It catalyzes the hydrolysis of glucose-6-phosphate to glucose and phosphate and is the only enzyme capable of forming significant amounts of glucose in the body. The deficiency of the catalytic subunit of G-6-Pase results in glycogen storage disease type Ia. Other subtypes of glycogen storage disease type I are presumably due to defects in the transport proteins. 5
GSD-Ia (von Gierke disease), caused by a deficiency of the G-6-Pase enzyme, is an autosomal recessive disorder of humans with an incidence of approximately 1:100,000 live births. Human patients with GSD-Ia commonly present at 3–4 months of age with failure to thrive, hepatomegaly, and/or hypoglycemic seizures. Biochemical abnormalities include hypoglycemia, lactic acidemia, hyperlipidemia, and hyperuricemia. 5 Current treatments are aimed at controlling symptomatic hypoglycemia by frequent feeding, continuous night-time feeding by a nasogastric tube, and/or oral administration of uncooked cornstarch. 4 7 10 These treatments have significantly alleviated the metabolic abnormalities of GSD Ia and improved the prognosis. However, the underlying pathological process remains untreated, and in adults, long-term complications can develop such as gout, hepatic adenomas with risk for malignancy, osteoporosis, progressive renal disease, delayed puberty, short stature, and pulmonary hypertension. 5
The catalytic subunit of human, canine, mouse, and rat liver G-6-Pase have been cloned. All four have an open reading frame of 1,071 nucleotides that code for a predicted 357 amino acid sequence. 12 14 20 23 The canine cDNA has also been cloned. 12 The gene structure and 5′ flanking sequence of human and rat G-6-Pase also have been sequenced. 1 23 Mutations in the G-6-Pase gene responsible for GSD-Ia in humans have been identified. 17–20 A G-6-Pase knockout mouse model has been described in the literature. These mice present with hypoglycemia, increased triglycerides, increased cholesterol, and uric acid. 16 The major limitations of this mouse model are the absence of lactic acidosis (an important feature of GSD Ia in humans), 5 their small size, and their limited survival, thus making study of long-term complications not feasible. Another limitation is the potential difference in response to viral gene therapy vectors between mice and larger mammals such as dogs. 12
We have previously reported the necropsy pathology findings of the first naturally occurring canine GSD Ia in the Maltese breed, 2 characterized a full-length cDNA encoding the canine G-6-Pase gene, and identified the disease-causing mutation in this model. 12 No clinical or laboratory data were accessible at that time. Due to the small size of the Maltese breed, the difficulty with survival of newborns (normal or affected), and the small litter size, we crossbred carrier Maltese with Beagles and established a breeding colony. In the present study, we describe the natural history, clinical and laboratory features, pathology, and complications of GSD Ia in this Maltese-Beagle model and also report the genomic structural organization of the canine G-6-Pase gene.
Materials and Methods
The experimental protocol was approved by the Institutional Animal Care and Use Committee at North Carolina State University. Animals were housed and work conducted in AALAC-approved facilities at NC State.
Breeding colony
Semen collected from a known Maltese GSD Ia carrier (P1) was diluted in an egg yolk-based extender and used to artificially inseminate three female Beagle dogs (P1). Twenty-one Maltese-Beagle crossbred pups (F1) were delivered from these breedings, including three males and nine females heterozygous for the mutated G-6-Pase gene. When sexually mature, three of the female Maltese-Beagle carriers (F1) were bred naturally or artificially inseminated with semen freshly collected from one of the male Maltese-Beagle carriers (F1). As a result of these three breedings, 10 pups (F2) were born or delivered by cesarian section. After birth, pups were weighed and photographed and blood was collected for determination of blood glucose concentration and GSD Ia DNA mutation analysis.
Puppies that were weak, hypoglycemic, or had hepatomegaly were given supplemental formula (Esbilac®, PetAg Inc., Hampshire, IL) containing 1.8 g polycose (Ross Products, Abbott Laboratories, Columbus, OH) at 1 ml/100 g body weight every 2 hours in 60 ml of formula in addition to nursing. The formula provided 23% of total calories as carbohydrate, 20% as protein, and 57% as fat. Once the diagnosis of GSD Ia was confirmed, the puppies were watched closely for signs of hypoglycemia. Feeding intervals were increased to 3 hours after 1 week and to a maximum interval of 4 hours after 3 weeks. Amounts fed at any given feeding were increased as tolerated to a maximum of 30 ml/100 g. At 6 weeks of age, cornstarch was introduced (0.9 g polycose and 0.9 g cornstarch in 60 ml Esbilac formula) to all puppies. At 6 weeks, affected puppies were also gradually started on solid feed using a commercial canine growth diet (Hill's® Science Diet Canine Growth, Hill's Pet Nutrition, Inc., Topeka, KS) that contained approximately 30% protein, 20% fat, and 45% carbohydrate on a dry-matter basis. From 4 months of age, pups were fed Esbilac supplemented with uncooked cornstarch only (1 g/kg in 60 ml Esbilac) at 4-hour intervals and solid food was continued.
Serum chemistry
Serum biochemical profiles were determined by an automated clinical chemistry analyzer (Instrumentation Labs Monarch 2000, Lexington, MA). Plasma lactate levels were determined using a dry chemical analyzer (Kodak DT60, Rochester, NY). Blood glucose levels in neonatal puppies were determined using a hand-held glucose analyzer (Accu-check Advantage, Boehringer Mannheim, Indianapolis, IN). Blood samples were obtained after a 1–2 hour fast at 1 and 4 weeks of age for determination of blood glucose, lactic acid, uric acid, cholesterol, and triglycerides. Complete biochemical profiles were obtained every 2–3 months thereafter. In addition to the above, blood samples for biochemical profiles were obtained from unaffected puppies at 6 and 8 weeks of age.
Pathology
Puppies that were euthanized or died were necropsied within 12 hours of death. Tissues were fixed in 10% buffered neutral formalin, embedded in paraffin, sectioned at 5–6 µm, and stained with hematoxylin and eosin. Three to 4 µm thick sections of kidney tissue from puppies D and E, which died at 32 and 60 days of age, respectively, were stained with periodic acid-methamine silver, periodic acid-Schiff with amylase digestion, trichrome, giemsa, and phosphotungstic acid hematoxylin stains. Liver tissues from stillborn puppies A and B and a littermate, puppy C, which died at 36 hours of age, were stained with periodic acid-Schiff to detect the presence of glycogen. Frozen sections of formalin-fixed liver tissue from the same puppies were examined for the presence of lipid using the oil-red-O stain.
Transmission electron microscopy
Formalin-fixed kidney tissue from puppy E, an affected puppy that died at 60 days of age, was additionally fixed in 1% osmium tetroxide/0.1 M phosphate buffer for 1 hour, dehydrated, infiltrated with Spurr resin, and polymerized at 70 C overnight. Ultrathin sections were prepared for examination with a Philips EM208S transmission electron microscope. 9
Enzyme and glycogen analysis
Aliquots of liver collected from two affected puppies (B and C) at necropsy were frozen at −70 C. Glycogen content was measured by complete digestion of polysaccharide using amyloglucosidase (Sigma Chemical Co., St. Louis, MO). The structure of the polysaccharide was inferred by using phosphorylase free of the debranching enzyme to measure the yield of glucose-1-phosphate. 3 27 Specific G-6-Pase activity was measured using G-6-Pase substrate after subtraction of nonspecific phosphatase activity as estimated by b-glycerophosphate. 8 Phosphorylase was measured by determination of inorganic phosphate released from 0.1 M G-1-phosphate in the presence of 1% glycogen and 2 mM adenosine monophosphate. 15
Canine genomic library screen
A canine genomic library (λ Dash II, Stratagene, Inc., La Jolla, CA) was screened by hybridization with a full-length canine G-6-Pase cDNA probe. The probe was radiolabeled with [α32P]dCTP using a Megaprime kit (Amersham Corp., Arlington Heights, IL). Phage was plated (5 × 104 pfu/150- × 15-mm plate) and duplicate nitrocellulose filters were lifted. Filters were subsequently prehybridized in 2× Pipes buffer, 50% deionized formamide, 0.5% sodium dodecyl sulfate (SDS), and 0.1 mg/ml sonicated salmon sperm DNA. Next, filters were hybridized at 42 C overnight in 6× sodium chloride and sodium citrate (SSC) buffer, 20 mm NaH2PO4, 0.4% SDS, and denatured, sonicated salmon sperm DNA. Filters were washed in 0.1× SSC and 0.1% SDS at room temperature and exposed to X-ray film for 24 hours. Phage was taken through six successive screening rounds. Five plaque-pure clones were obtained that contained overlapping segments of the G-6-Pase gene.
Phage preparation and purification
Stock lambda phage solutions of the screened clones were prepared (plate lysate protocol II) and liquid lysates were obtained using the infection at low multiplicity protocol described in Sambrook, et al. 21 Phage DNA was purified using both the Lambda Wizard Preps DNA Purification System (Promega, Madison, WI) and the Qiagen Lambda Kit (Qiagen, Chatsworth, CA). The concentration of purified clones was approximated by running samples on agarose gels flanked with Low DNA Mass Ladder (Life Technologies, Gaithersburg, MD).
DNA sequencing and analysis
Direct sequencing of five purified G-6-Pase clones was accomplished on an Applied Biosystems 377 Automated Sequencer (ABI, Foster City, CA) using fluorescent-tagged dRhodamine dye-terminators (dRhodamine Terminator Cycle Sequencing Kit, Foster City, CA) utilizing the T3 and T7 primers found in the λ Dash II polylinker. All five clones were sequenced using sense and antisense primers that span the G-6-Pase gene. Exon/intron boundaries were confirmed by sequencing across the putative junction from within the intron. Sequence data were analyzed using the GCG sequence software package (UNIX, version 9, Genetics Computer Group, Madison, WI).
Mutational analysis
Canine genomic DNA samples were screened for the M121 mutation by restriction enzyme analysis, as previously described. 12 Genomic DNA was purified from either bloodspots (methanol wash/ethanol precipitation) or whole blood (Puregene DNA Isolation Kit, Gentra Systems, Minneapolis, MN). A genomic DNA fragment was amplified with primers flanking the mutation, digested with Nco1, subjected to electrophoresis on a 2% Nusieve agarose gel, and repeated in all cases to confirm findings. Loss of a Nco1 restriction site in both alleles was seen in affected puppies.
Results
Breeding colony
The six affected puppies described in this study were from three litters of Maltese-Beagle carrier breedings totaling 10 puppies (Table 1). Two puppies were stillborn (A and B) and one died at 2 days of age (C) despite being fed by nasogastric tube. One of the other affected puppies survived for 32 days (D) before euthanasia due to respiratory distress. Another affected puppy (E) was euthanized at 60 days of age following the acute onset of dyspnea suspected to be caused by aspiration pneumonia. A sixth affected puppy (F) is still alive at 15 months. Of the remaining four puppies, two were carriers and two were normal. The affected puppies that survived were hypoglycemic at birth, with blood glucose values <40 mg/dl compared with 81–126 mg/dl for normal and carrier puppies.
Summary of findings of affected puppies.

At birth, all affected puppies had profound abdominal distention due to hepatomegaly. Puppies C, D, and E, which survived the immediate perinatal period, had persistent hepatomegaly based on serial physical examinations and abdominal ultrasonography. All three puppies were weaned early because of failure to nurse, and the subsequent development of hypoglycemia with associated weakness, obtunded mental state, and seizures was seen. The single living affected puppy (F) has persistent hepatomegaly with increased echogenicity throughout the hepatic parenchyma, consistent with glycogen and fat deposition. This puppy also developed enlarged kidneys at 3–4 months of age and had multiple choleliths at 6–7 months of age.
Additional abnormalities noted in affected puppies included poor growth rates (Fig. 1) plus delayed development of neurological reflexes, opening of the eyes, visual development, and weaning to solid food. Biochemical abnormalities in affected puppies parallel those seen in affected humans and include hypoglycemia, hyperlactacidemia, hyperuricemia, hypercholesterolemia, and hypertriglyceridemia (Tables 2,3).
Select biochemical data from puppy F.∗
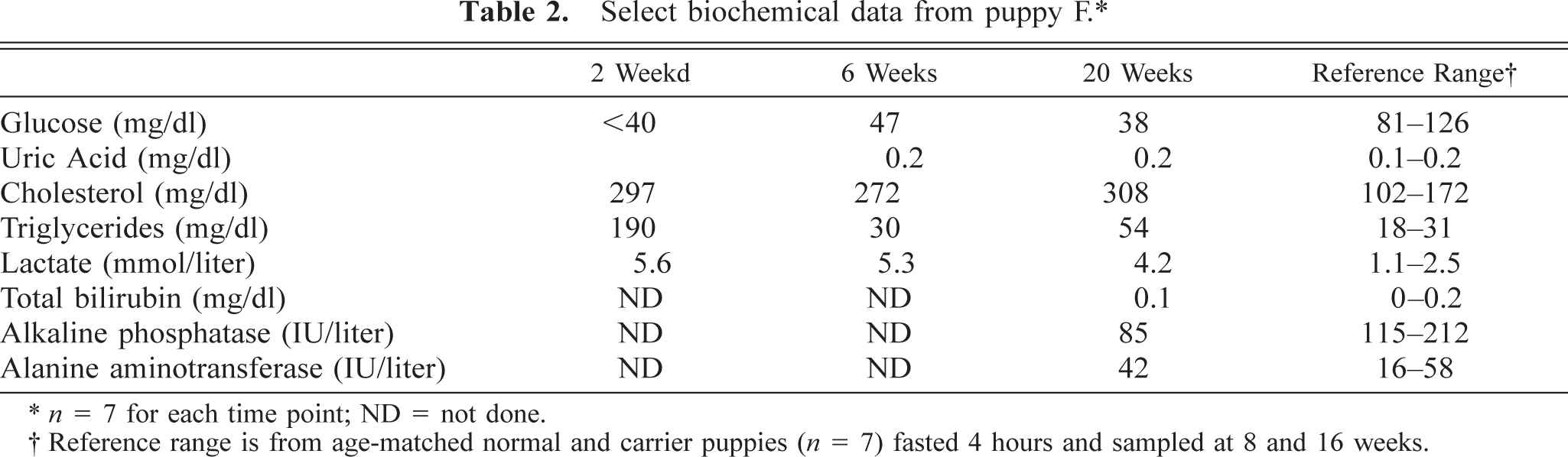
n = 7 for each time point; ND = not done.
Reference range is from age-matched normal and carrier puppies (n = 7) fasted 4 hours and sampled at 8 and 16 weeks.
Select biochemical data from puppies D and E.∗
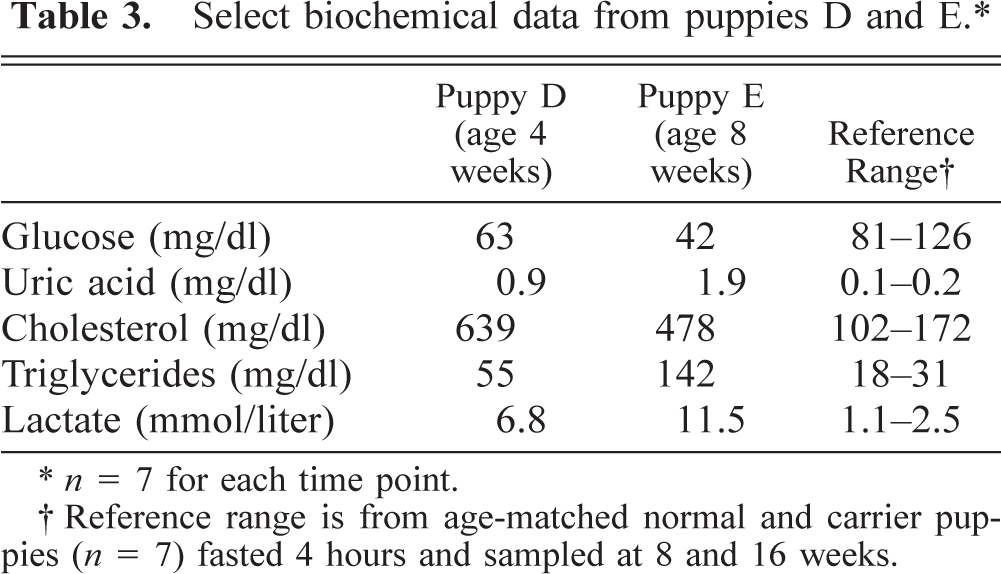
n = 7 for each time point.
Reference range is from age-matched normal and carrier puppies (n = 7) fasted 4 hours and sampled at 8 and 16 weeks.

An affected puppy compared with normal littermate. An affected puppy (left), weight 380 g, is compared with a normal littermate (right), weight 660 g, at 14 days of age. Notice the abdominal distention in the affected puppy.
Pathology
Five affected puppies were necropsied. The livers of all puppies except puppy E were mildly to moderately enlarged and pale. The liver of puppy E, which died at 60 days of age, was markedly enlarged, pale tan, and friable. Additionally, there was mild dilation of the lateral ventricles in the brain of puppy E. Microscopically, the hepatocytes in all the affected puppies were markedly vacuolated, causing compression of sinusoids and a loss of the normal lobular hepatocellular architecture (Fig. 2). Glycogen and lipid were demonstrated in vacuolated hepatocytes of puppies A, B, and C using the periodic acid-Schiff and oil-red-O stains. The proximal convoluted tubules of affected puppies also were mild to moderately vacuolated, suggesting the accumulation of glycogen. In the kidneys of puppies D and E, there was moderate to marked glomerular sclerosis characterized by focal segmental to diffuse increased cellularity and granularity of many glomerular tufts (Fig. 3). Further characterization of the glomerular lesion was not possible using periodic acid-methamine sliver, periodic acid-Schiff, trichrome, and giemsa stains. Renal cortical tissue from puppy E was examined by transmission electron microscopy. The basement membranes of glomerular capillaries were of uniform thickness and interpreted to be normal. Black granules, suggestive of accumulated glycogen, were present in some areas of the mesangial matrix.

Liver; affected puppy C, which died at 2 days of age. Marked, diffuse vacuolation of hepatocytes causing sinusoidal compression and marked distortion of normal hepatocellular architecture. Clear vacuoles in hepatocytes contained glycogen and lipid. H&E. Bar = 125 µm.
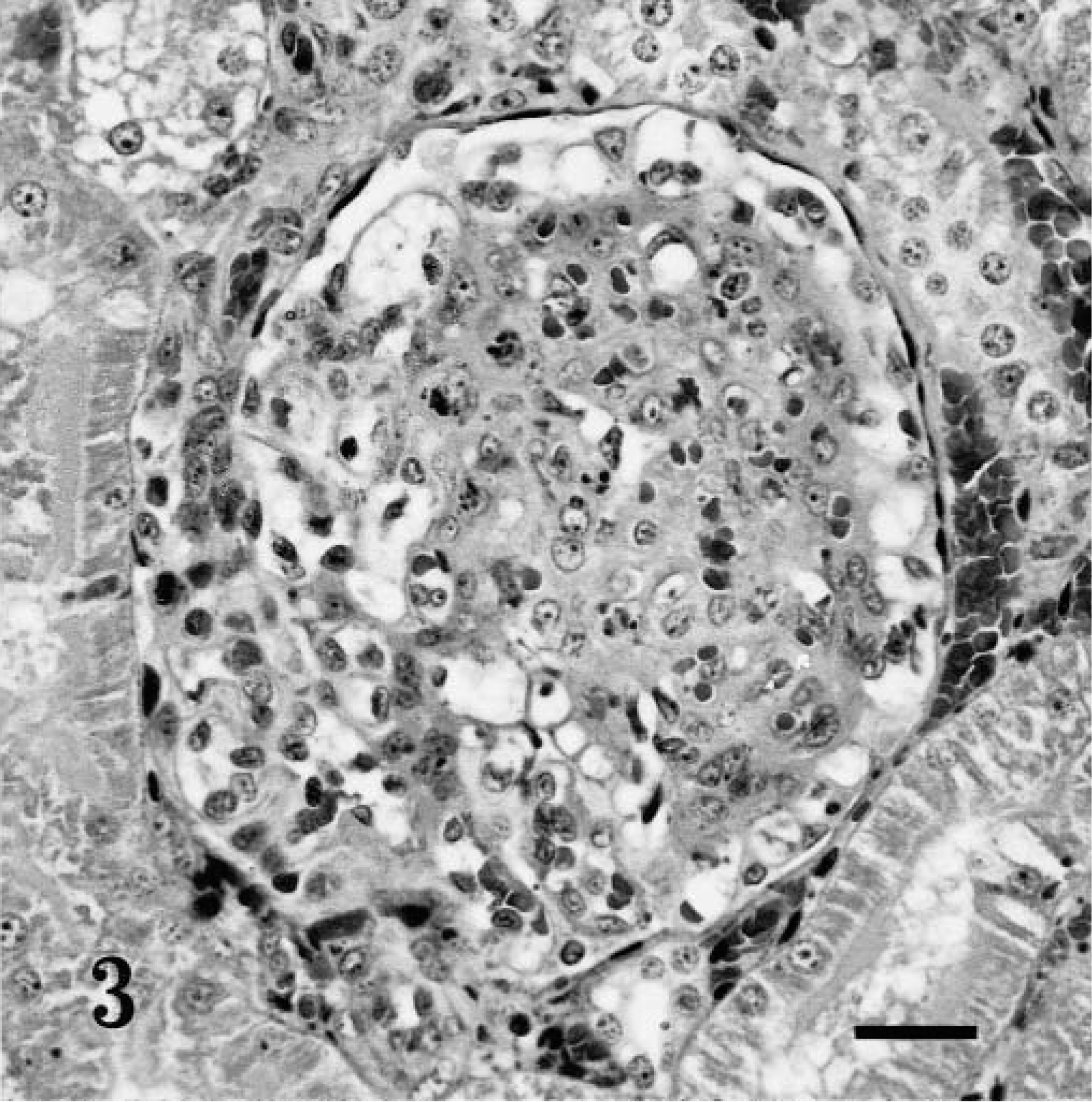
Kidney; affected puppy E, which died at 60 days of age. Focal, segmental glomerular sclerosis. Phosphotungstic acid hematoxylin stain. Bar = 100 µm.
Enzyme assays and glycogen analysis
Results of enzyme assays in puppies B and C revealed isolated, markedly reduced levels of G-6-Pase in their livers. G-6-Pase levels varied between 0.5 and 0.7 mmol/min/g tissue in these two puppies, compared with 8 mmol/min/g tissue in age-matched control puppies. Hepatic values of phosphorylase in affected puppies were similar to values in control puppies. The glycogen content in the livers was between 6.9 and 9.6% in affected puppies (B and C), compared with <6% in control puppies.
Genomic structure organization
The canine G-6-Pase gene was isolated from a canine genomic λ Dash II phage library. Five plaque-pure clones were isolated that had overlapping sequences. The canine G-6-Pase gene is approximately 11.8 kb in length and contains five exons. The exon–intron structure of the gene is shown schematically (Fig. 4). All boundaries conform to intron consensus criteria [gt(a/g) … pyrimidine rich … ag] and are conserved in the human, rat, mouse, and canine G-6-Pase genes. 1 20 23

Exon/intron structure demonstrating exon/intron boundaries for the canine glucose-6-phosphatase gene. Twenty-five base pairs of intronic sequence are shown for each of the five exon/intron boundaries. Consensus sequences for 5′-donor and 3′-acceptor sequences are also shown. Sizes of respective exons and introns are also given.
The first 1.5 kb of the 5′ region of the canine G-6-Pase gene was isolated and sequenced in both directions. The sequence is shown in Fig. 5. The 5′ region contained a consensus sequence for a TATAA-box located at position −39 and a consensus sequence for a CCAAT box located at position −116. There are a number of sites with sequence homology to putative binding sites for liver-enriched transcription factors in the 5′ region of human and rat sequence. These putative response element motifs are as follows: GRE (glucocorticoid response element), positions −796 (93% similarity), −313; IRS (insulin response sequence), position −820 (90% similarity); HNF1 and HNF5 (hepatocyte nuclear factor), positions −584, −368, −237, −150; LF-A1 (liver-specific transcription factor), positions −1346, −1288; C/EBP (CCAAT enhancer binding protein), positions −784, −173; CRE (cAMP response element), position −173; and AP1 and AP2 (activating protein), positions −485, −459, −209 (87% similarity). These response elements are shown in Fig. 5. Many of the putative binding sites in the 5′ promoter region of the canine G-6-Pase gene are similar/identical to those in the human and rat 5′ promoter region. The presence and clustering of these sites suggests that this may be a highly regulated portion of the 5′ region of the gene.
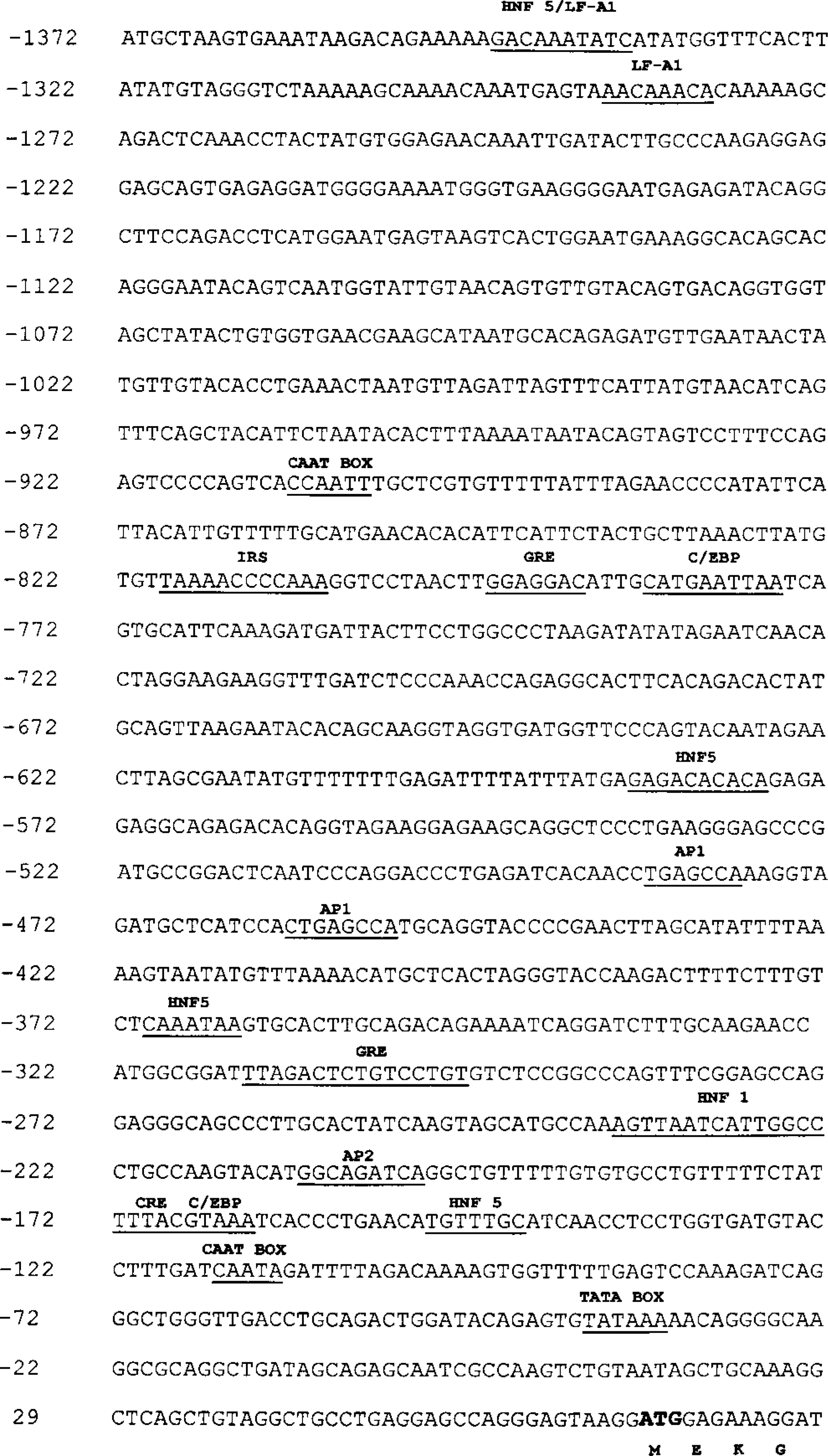
Sequence of the first 1.5 kb of 5′-flanking region of the canine glucose-6-phosphatase gene. Various putative response element motifs are underlined and identified as follows: glucocorticoid response element (GRE), −796, −313; hepatocyte nuclear factor (HNF), −584, −368, −237, −150; liver-specific transcription factor (LF-A1), −1346, −1288; insulin response sequence (IRS), −820; cAMP response element (CRE), −173; CCAAT enhancer binding protein (C/EBP), −784, −173; activating protein (AP), −485, −459, −209. Putative TATA (position −39) and CAAT (position −116 and −910).
Discussion
We have established a breeding colony of carrier and affected dogs with glycogen storage disease type Ia and report the clinical features and natural history of the disease in this animal model. The clinical features in the affected puppies of postnatal onset of failure to thrive, delayed growth, moderate to marked hepatomegaly, fasting hypoglycemia, lactic acidosis, hyperuricemia, and hypertriglyceridemia closely mimic human disease. Affected puppies require intensive support and frequent feeding and are profoundly susceptible to stress-induced hypoglycemia, representing a dramatic phenotypic expression of GSD Ia in this animal model.
The gross and microscopic appearance of the liver lesions are consistent with those that have been previously described in two Maltese puppies, a mouse model, and humans with GSD Ia. 2 5 16 Hepatocellular vacuolation was present in stillborn, affected puppies suggesting that the effect of G-6-Pase deficiency begins in utero and needs to be further evaluated. The focal segmental glomerulosclerotic lesions in puppies D and E that died at 32 and 60 days of age, respectively, have not been described in G-6-Pase-deficient Maltese puppies or in the mouse model of glycogen storage disease type Ia. However, morphologically similar glomerulosclerotic lesions have been described in children and young adults with glycogen storage disease type Ia. 6 25 Ultrastructural lesions in kidney specimens from G-6-Pase-deficient children include marked increase in mesangial matrix, irregular thickening of glomerular basement membranes, and increased glycogen deposition in the mesangium and within mesangial, epithelial, and endothelial cells. 6 25 Ultrastructural examination of formalin-fixed tissue from puppy E was inconclusive. Evidence of glomerular basement membrane thickening was not apparent, but the mesangial matrix was dense and contained dark granules suggestive of glycogen accumulation. Further study of the glomerular lesions is warranted to determine the similarities to renal lesions in human patients with glycogen storage disease type Ia and to assess the potential of using the Maltese-Beagle cross model to study the pathogenesis of renal disease caused by G-6-Pase deficiency.
The mild hydrocephalus in puppy E was interpreted to be an incidental finding unrelated to G-6-Pase deficiency. Congenital hydrocephalus occurs occasionally in toy breeds of dogs, including the Maltese breed, and often is an incidental finding at necropsy when death is unrelated to neurological dysfunction. 24 Hepatocellular adenomas have been reported in human patients with GSD Ia, with an increase in incidence after puberty. 5 To date, there has been no gross or microscopic evidence to suggest similar lesions in affected puppies. This could be due to the younger age of affected puppies studied to date. A longer term observation of affected puppies will thus be necessary to determine if hepatocellular adenomas are part of the phenotypic expression of GSD Ia in this animal model.
Current therapy for GSD Ia consists of sufficient carbohydrate supplementation, either by uncooked cornstarch or continuous glucose infusion, to prevent hypoglycemia. Uncooked cornstarch, dosed at 1.5–2.5 g/kg every 4–6 hours, provides a slow-release form of glucose supplementation; however, treatment in infancy can require continuous nasogastric tube feeding of glucose due to starch intolerance. These treatments have clearly improved the outcome of GSD Ia; however, neither treatment prevents hypoglycemia and lactic acidosis entirely, and nasogastric feeding has the accompanying risk of life-threatening equipment failure. 5 Biochemical monitoring of 17 children and young adults who were treated with nutritional therapy for GSD I demonstrated early morning hypoglycemia (24%) and elevated lactic acid (88%), despite compliance with cornstarch treatment. These 17 patients had decreased average height, increased average weight, anemia (35%), hepatic adenomas (29%), glomerular hyperfiltration (94%), and increased urinary albumin excretion (12%) after documented compliant nutritional therapy for almost 13years. 26 Thus, long-term complications do occur in patients who adhere to the currently available treatments.
Compliance is another issue as children enter adolescence. The restrictive nature of the diet and the side effects of cornstarch, such as weight gain and abdominal bloating, are all contributing factors to noncompliance. Nutritional deficiencies have been recently reported as another complication of the disease. 13 Hence, current therapy for GSD Ia is inadequate to prevent many of the long-term complications of this disorder. The availability of animal models with the disease would greatly facilitate more novel therapeutic approaches such as drug and genetic replacement therapy.
The recent demonstration of a correction of abnormal plasma glucose, cholesterol, and triglyceride profiles and decrease in liver and kidney size in G-6-Pase-deficient mice treated with a single administration of Ad-mG-6-Pase, a second generation recombinant adenovirus containing the murine G-6Pase cDNA, suggest that gene therapy with adenovirus vectors could provide at least temporary resolution of GSD Ia. 28 However, the marked decrease in G-6-Pase levels 2 months following vector administration in the mice and the concerns regarding hepatic toxicity of adenovirus vectors reinforces the need to improve current viral vectors prior to considering clinical applications. The availability of the mouse and canine model of GSD Ia would thus definitely facilitate thorough evaluation of safety and efficacy of gene therapy protocols prior to any human clinical trials.
In addition to the cDNA, we have isolated and sequenced the first 1.5 kb of the 5′ region of the canine G-6-Pase gene, which contains key putative regulatory elements. DNA vectors can now be developed to treat GSD Ia in this canine model through liver-targeted delivery and hepatic expression of G-6-Pase in the neonatal period. Furthermore, studies can be done using vectors that contain the G-6-Pase promoter in addition to cDNA to see if it induces more regulated, tissue-specific expression of G-6-Pase driven by the G-6-phosphate promoter.
Establishment of this GSD Ia canine colony, with affected dogs expressing clinical signs, biochemical abnormalities, and pathological changes, closely resembling the human disease and isolation of the canine G-6-Pase gene, provides a means to follow the natural history, study the pathophysiology, and test newer treatments such as drug and genetic therapy.
Footnotes
Acknowledgements
This work was supported in part by M01-RR-30, National Center for Research Resources, General Clinical Research Program (PK is a CAP), the Raleigh Kennel club, and The Childrens Miracle Network Telethon Grant. We thank Ms. Barbara Merrick and Dr. Diane Egnor for their expertise in establishing the initial Maltese colony. We would also like to thank Ms. Deborah Lasater for preparation of the manuscript.