
Abstract
Mice with null mutations of ciliary neurotrophic factor (Cntf) receptor alpha (Cntf-Rα), or cytokine-like factor 1 (Clf), one component of Cntf-II (a heterodimeric Cntf-Rα ligand), die as neonates from motor neuron loss affecting the facial nucleus and ventral horn of the lumbar spinal cord. Exposure to cardiotrophin-like cytokine (Clc), the other putative Cntf-II element, supports motor neuron survival in vitro and in ovo. Confirmation that Clc ablation induces an equivalent phenotype to Clf deletion would support a role for Clc in the functional Cntf-II complex. In this study, Clc knockout mice had decreased facial motility, did not suckle, died within 24 hours, and had 32% and 29% fewer motor neurons in the facial nucleus and lumbar ventral horn, respectively; thus, Clc is essential for motor neuron survival during development. The concordance of the Clc knockout phenotype with those of mice lacking Cntf-Rα or Clf bolsters the hypothesis that Clc participates in Cntf-II.
Ciliary neurotrophic factor (CNTF) is a neuropoietic cytokine that supports neuron survival in the central nervous system and thus has been touted as a potential therapeutic agent for ameliorating neurodegenerative diseases. 7 However, 2 lines of evidence call into question the premise that CNTF therapy will afford significant improvement in patients with neurodegenerative conditions. First, a large fraction of the Japanese population (approximately 2.5%) are homozygous for an inactivating mutation of the CNTF gene and yet do not develop an increased incidence of neurologic disease with age. 9 Second, mice lacking the Cntf receptor alpha (Cntf-Rα) die soon after birth because of defective suckling arising from a profound loss of motor neurons in the facial nucleus, 3 whereas mice with null mutations of Cntf are essentially normal. 3 Taken together, these data indicate that another CNTF-like ligand (designated CNTF-II) must exist that supports motor neuron survival in some or many CNTF-dependent neural domains during development, thus suggesting that CNTF therapy alone will be insufficient to slow or halt neurodegenerative diseases.
Recent evidence suggests that CNTF-II is a heterogeneous complex formed by the interaction of cytokine-like factor 1 (CLF; also called nodal-related protein 6 [NR6]) with an interleukin-6 (IL-6) family member known as cardiotrophin-like cytokine (CLC; also designated B cell–stimulating factor 3 [BSF-3] and novel neurotrophin 1 [NNT-1]). 4, 8 As with Cntf-Rα null mutant mice, animals lacking Clf die within 24 hours of birth because of defective suckling 1 and have reduced numbers of motor neurons in the facial nucleus and ventral horn of the lumbar spinal cord. 5 Treatment with exogenous CLC is known to support survival of chick and mammalian motor neurons in vitro 5, 6, 8 and to reduce the extent of physiologic apoptosis during neurogenesis in wild-type chick embryos in ovo. 5 However, the functions of CLC have not been confirmed in vivo in a gene-targeting experiment designed to see whether or not the phenotype of Clc null mutant mice recapitulates the phenotypes induced by ablation of either its putative coligand, Clf, or its proposed receptor, Cntf-Rα. In this brief communication, we describe the outcome of such a CLC knockout experiment.
The mouse Clc gene is encoded within 3 exons spanning more than 8,000 base pairs (8 kbp) of genomic DNA. A positive-negative selection replacement–type vector, pAmgenKO-KHJ-1, was designed that would replace the second and third exons with a PGKneo cassette (Fig. 1). Polymerase chain reaction (PCR) amplification was employed to generate 1.8-kbp short arm and 7.8-kbp long arm DNA fragments with homology to the 3′ and 5′ regions, respectively, of the mouse BSF-3/CLC/NNT-1 genomic locus. The resulting 1.8-kbp fragment contained restriction enzyme sites for Sgfl (5′ end) and AscI (3′ end), whereas the 7.8-kbp fragment bore sites for NotI (5′ end) and SalI (3′ end). Each PCR fragment was digested along with pAmgenKO (an Amgen proprietary vector) with the appropriate restriction enzymes, purified, and then ligated to the pAmgenKO vector. After expansion, the purified pAmgenKO-KHJ-1 targeting vector was introduced into 129/SvJ embryonic stem cells, which were then injected into C57BL/6J blastocysts. The blastocysts were transferred into pseudopregnant Tac:SW (Swiss Webster) females, after which chimeric (agouti) offspring were mated to Tac:N:NIH(S)-BC (Black Swiss) mice at 6–8 weeks of age to produce Clc heterozygous (+/− or Het) individuals. Young adult (8–14-week-old) Het parents were mated to generate wild-type (Clc +/+ or WT), Clc Het, and Clc homozygous knockout (−/− or KO) progeny.
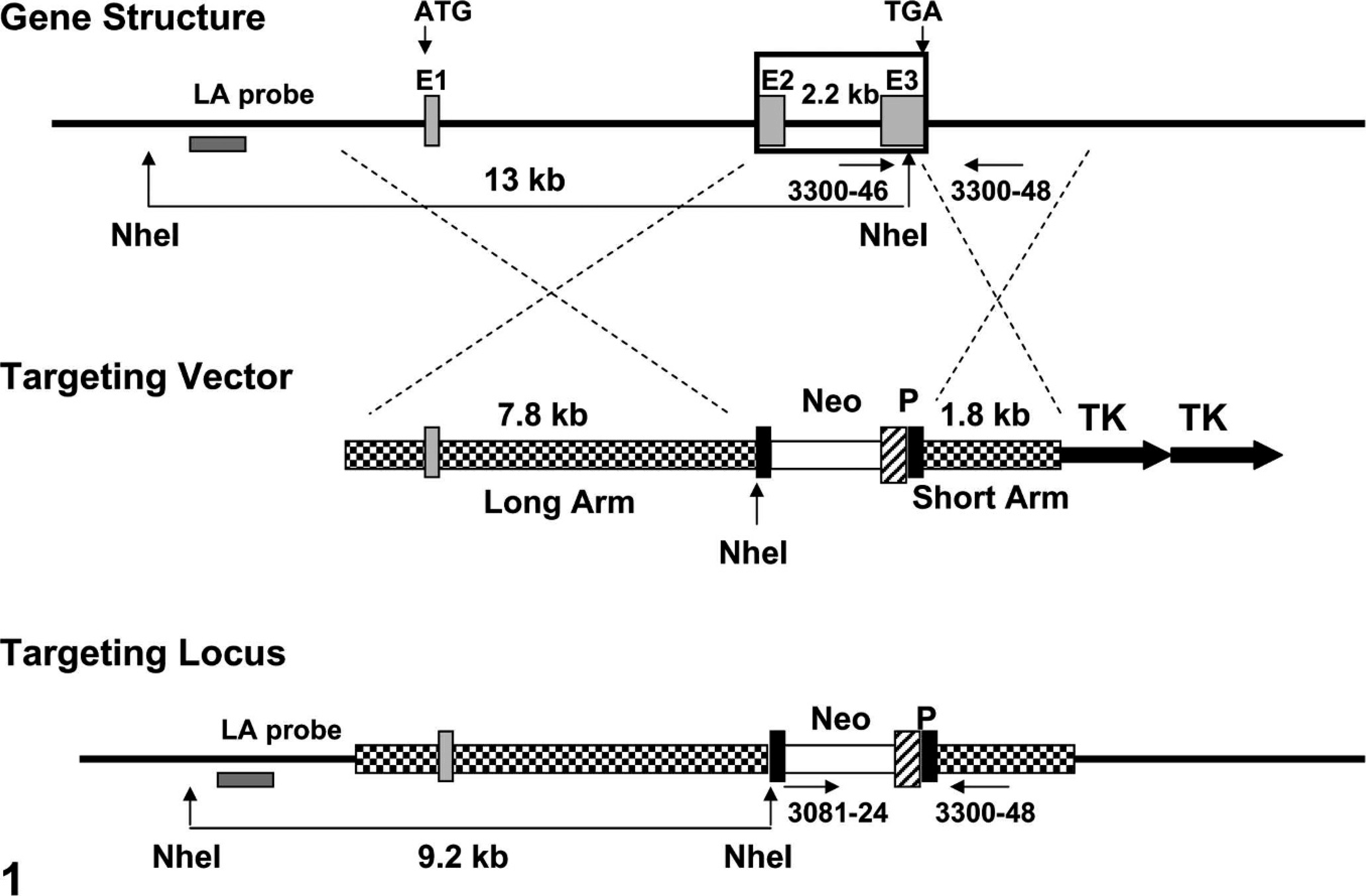
Targeting of the mouse cardiotrophin-like cytokine (Clc) gene. A 2.2-kbp span encompassing exons 2 (E2) and 3 (E3) (large rectangular box in top sequence) was replaced by a PGK-neo cassette (Neo) with the use of a proprietary vector, pAmgenKO-KHJ-1 (middle sequence). Abbreviations: ATG = start codon; LA = long arm; P = phosphoglycerate kinase (PGK) promoter (to drive Neo gene expression); TGA = stop codon; TK = thymidine kinase (gene permitting negative selection in cultured embryonic stem [ES] cells). The LA probe allows ES cells to be screened by Southern blot. Gene and targeting vector sequences bounded by the 21-mer PCR primers are: 3081-24, AGC AGC CTC TGT TCC ACA TAC A; 3300-46, CAA GGA CTT CAA CCG GCT TAA G; and 3300-48, CTC GAT GCC CAC TGT ACG ATT T. These primer sets define PCR products of 363 bp for the engineered Clc null mutant allele (by the 3081-24/3300-48 pair) or 289 bp for the wild-type allele (by the 3300-46/3300-48 pair).
Eleven litters from the Het × Het pairings were delivered in which appropriate maternal care was rendered to the neonates and in which all pups survived for the first 12 hours. Offspring were evaluated visually on an hourly basis during the first 36 hours after birth. Neonates were briefly removed from the litter, examined for normal facial movements, weighed, and killed by decapitation when they became moribund or at 36 hours after birth (whichever came first). Animals were weighed and tails harvested to allow DNA extraction. Genotyping was done by standard PCR methods with the use of matched primer sets (Table 1). A representative genotyping gel is shown in Fig. 2.
Primer sequences for genotyping.

∗ Identical to the upstream PCR primer designated 3300–46 (Fig. 1).
† Sequences that bracket the PCR primer designated 3300–48 (Fig. 1).
‡ Identical to the upstream PCR primer for the neo insert designated 3081-24 (Fig. 1).
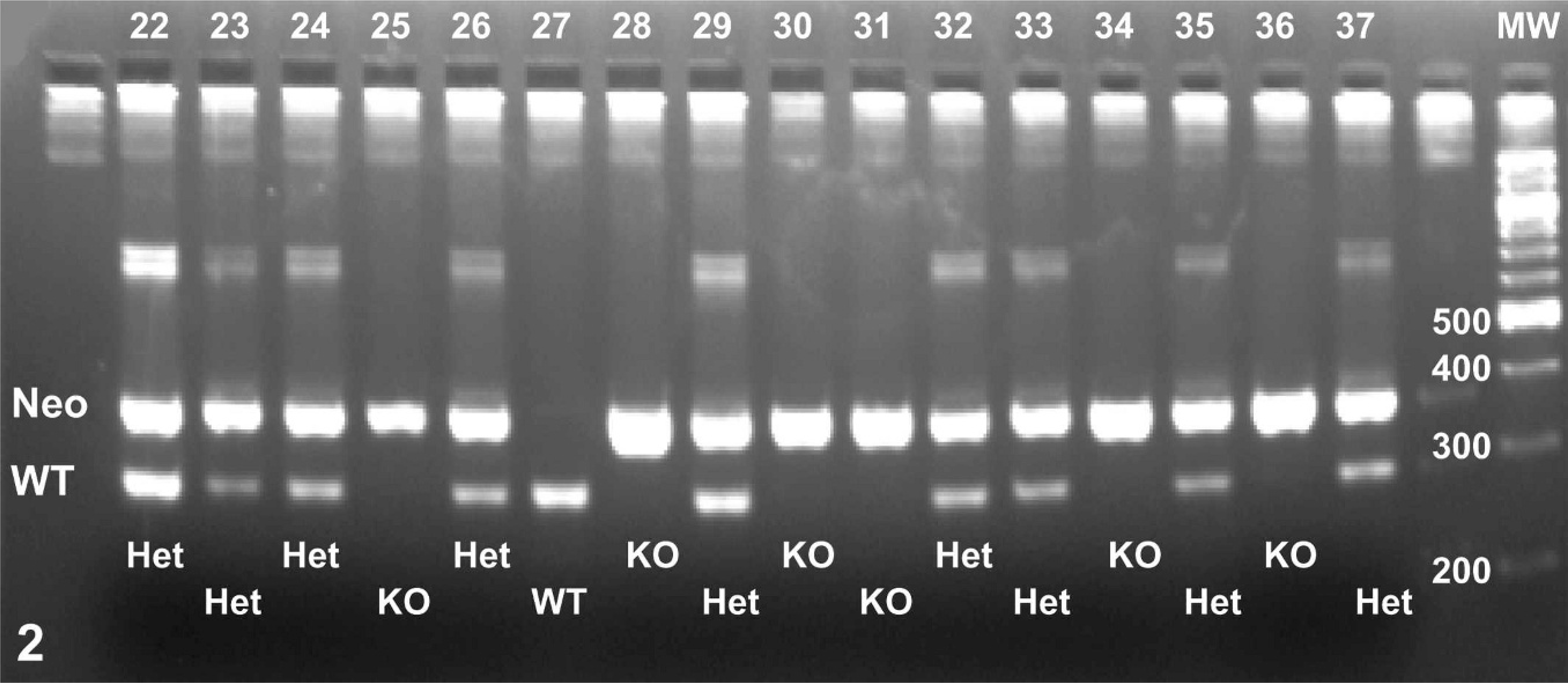
A representative genotyping gel showing that newly born litters had many Clc knockout (KO, 363-bp neo band only) pups as well as Clc wild-type (WT, 289-bp WT band only) and Clc heterozygous (Het, both bands) animals. The numbers across the top of the lanes denote individual pups from 3 distinct litters. The abbreviation “MW” over the far right lane identifies the molecular weight standard; the numbers adjacent to bands within the MW lane give the weight of the molecule composing the band.
Neonates were fixed by immersion in Bouin's solution or neutral buffered 10% formalin immediately on death or (after decapitation) at 36 hours after delivery. Following fixation, tissue blocks of selected WT and KO animals that contained the brainstem (for the facial nucleus) and lumbar vertebral column (for spinal motor neurons and dorsal root ganglia) were obtained with the use of standard anatomic landmarks, dehydrated in graded alcohols, cleared in Propar, and embedded in paraffin. One cassette was processed for each neonate. Tissues were cut at 4 μm, taking 1 or more sections at 40-μm intervals until 20 levels had been harvested. One section per level was stained with cresyl violet (to reveal large neurons). Neuron numbers were counted in 2 neuronal populations that are adversely affected by disruption of Cntf-Rα–mediated signaling (facial nucleus and ventral horn of the lumbar spinal cord 3 ), as well as in the dorsal root ganglia (a site unlikely to be affected 3 ). Manual counts of neuron cell bodies containing nuclei were made in 5 sections per site according to established criteria 11 on a bright field microscope equipped with a 10 × 10 square ocular grid. Counts were assessed by the nonparametric Wilcoxon rank sum test with commercially available statistical software (JMP v.5.0; SAS Institute, Cary, NC) with significance assigned to P ≤ .05.
The additional sections from WT neonates were used to evaluate neural Clc expression by isotopic in situ hybridization (ISH). Riboprobes were generated by transcription from a cDNA construct of the mouse Clc gene corresponding to nucleotides 1–787 (GenBank AF176913). A 33P-labeled RNA probe was made with T7 RNA polymerase after linearization with the KpnI restriction enzyme. A separate clone in the reverse orientation was used for generating the sense probe. A standard ISH protocol was followed, including proteinase K treatment, acetylation, and overnight hybridization at 60°C in a hybridization solution containing 1 × 106 cpm of 33P-labeled riboprobe per slide. After hybridization, slides were treated to RNAse digestion followed by a series of SSC washes with highest stringency of 0.1× SSC at 55°C for 30 minutes. The slides were coated with Kodak NTB2 emulsion and exposed for 3 weeks in the dark at 4°C, developed, and then counterstained with HE. The distribution and strength of Clc expression was evaluated by dark field microscopy and the use of a semiquantitative grading scale (absent, minimal, mild, moderate, or marked).
A total of 94 neonates were obtained from the 11 viable litters (n = 8.6 ± 0.8 pups [mean ± SEM] per litter; range, 5–12). Of these, 20 (21%) were WT, 61 were Het (65%), and 13 were KO (14%). All 13 (100%) KO and 3 (5%) Het neonates became moribund during the first 24 hours after birth. Relative to their littermates, KO mice had decreased facial motility on stimulation of the snout and did not suckle (as indicated by empty stomachs; Fig. 3). Absolute body weights of KO animals (mean ± SEM, 1.17 ± 0.03) were significantly decreased (P ≤ .05) at P1 by 25% relative to those of age-matched wild-type (1.57 ± 0.03) and Het (1.61 ± 0.04) littermates. No other gross abnormalities were evident in engineered mice.
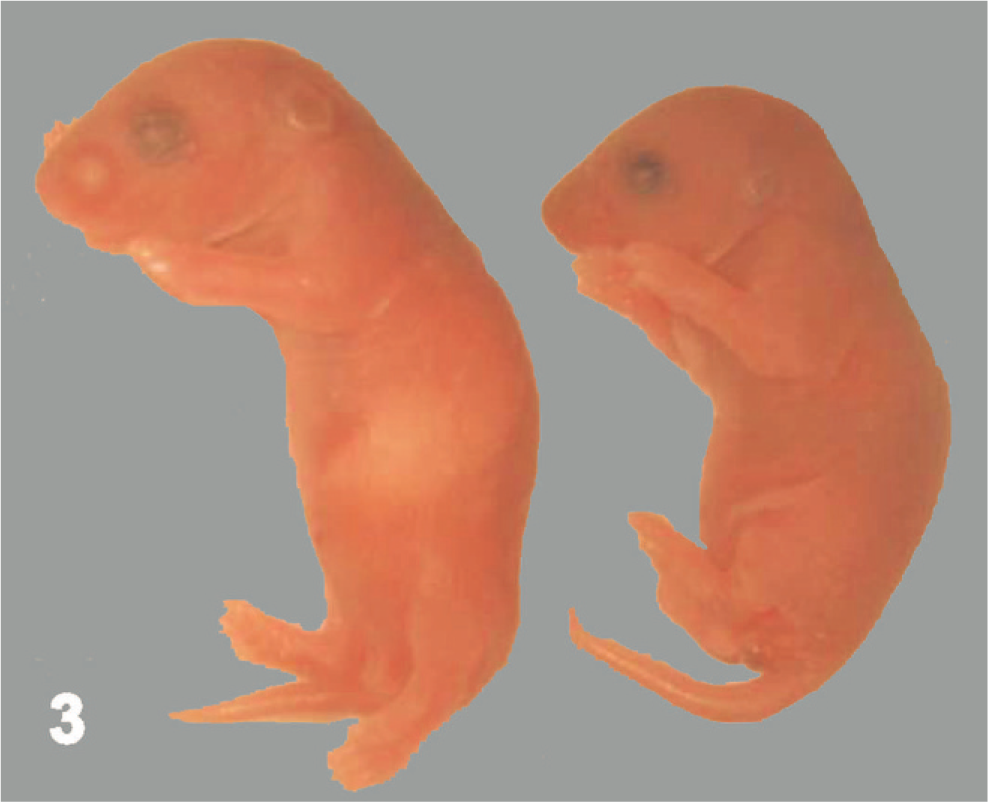
Neonatal mice (postnatal day 1). Relative to its Clc wild-type littermate (left), the Clc knockout pup (right) has a smaller profile and a shrunken stomach (indicated by wrinkled skin over the ventral torso and the lack of a milk spot).
Overt gross or histopathologic abnormalities were not apparent in neural tissues of KO neonates (including cerebrum, midbrain, cerebellum, brainstem, spinal cord, cranial and dorsal root ganglia [DRG], cranial nerves, and spinal nerve roots) during qualitative assessments. However, absolute counts of facial nucleus motor neurons and spinal motor neurons were reduced significantly (P ≤ .05) in KO neonates by 32% and 29%, respectively, relative to numbers in WT littermates (Table 2). Counts in the DRG were not affected in KO mice.
Neuron count data.
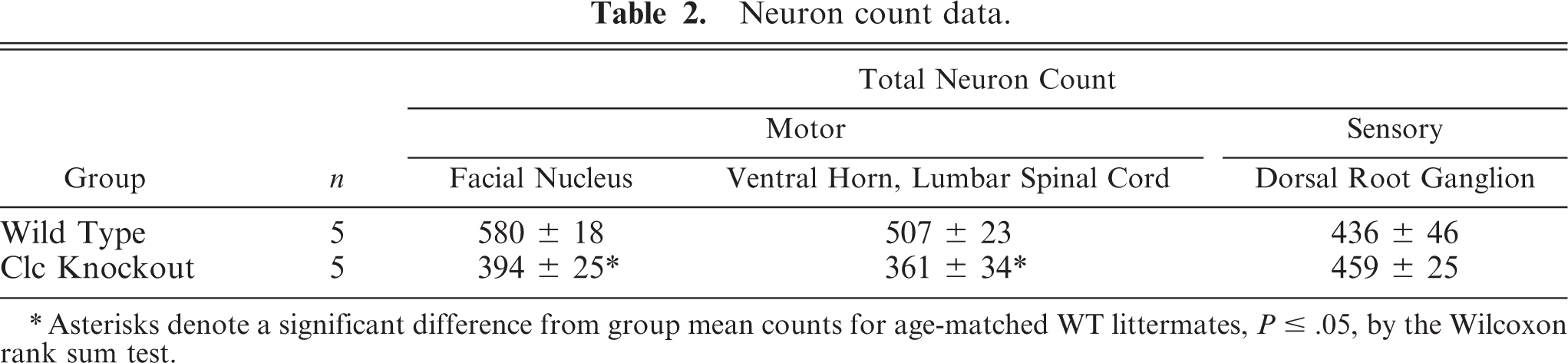
∗ Asterisks denote a significant difference from group mean counts for age-matched WT littermates, P ≥ .05, by the Wilcoxon rank sum test.
Neuronal expression of Clc in WT neonates (postnatal day 1) was absent in motor neurons of the facial nucleus (the nucleus that controls suckling in neonates 11 ; Fig. 4) but was evident in mild amounts in the sensory neurons of the neighboring cochlear nucleus (Fig. 5). Interestingly, a minimal signal was seen in the dorsal root ganglia in neonates (i.e., a neuronal cell population shown to be unaffected by null mutations of Cntf-Rα, 3 Clf, 5 or Clc; Table 2; Fig. 6).
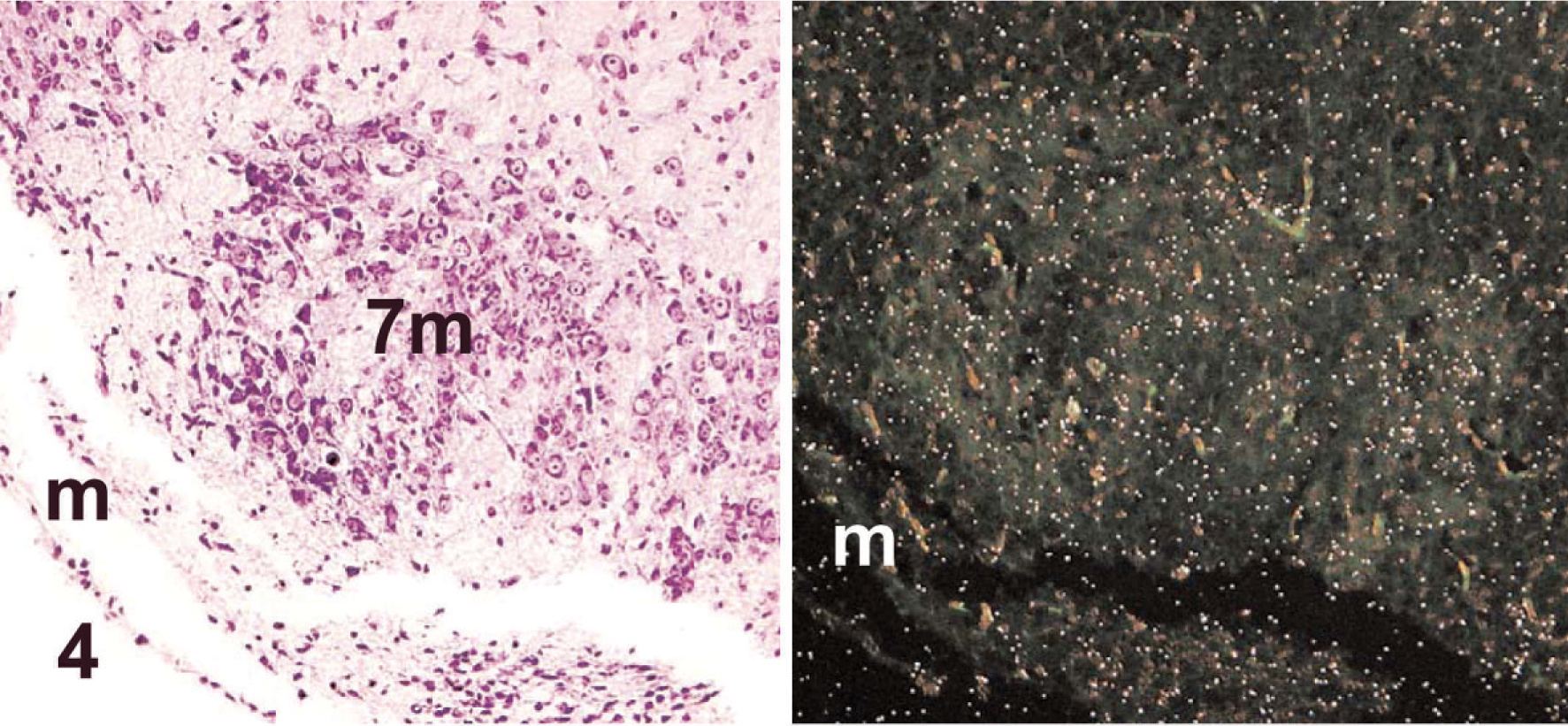
Brainstem; wild-type mouse neonate (postnatal day 1). Motor neurons of the facial nucleus (7m) do not express Clc. The meninges (m) are identified for orientation. Stains: HE (left), isotopic in situ hybridization (anti-sense) for Clc (right).
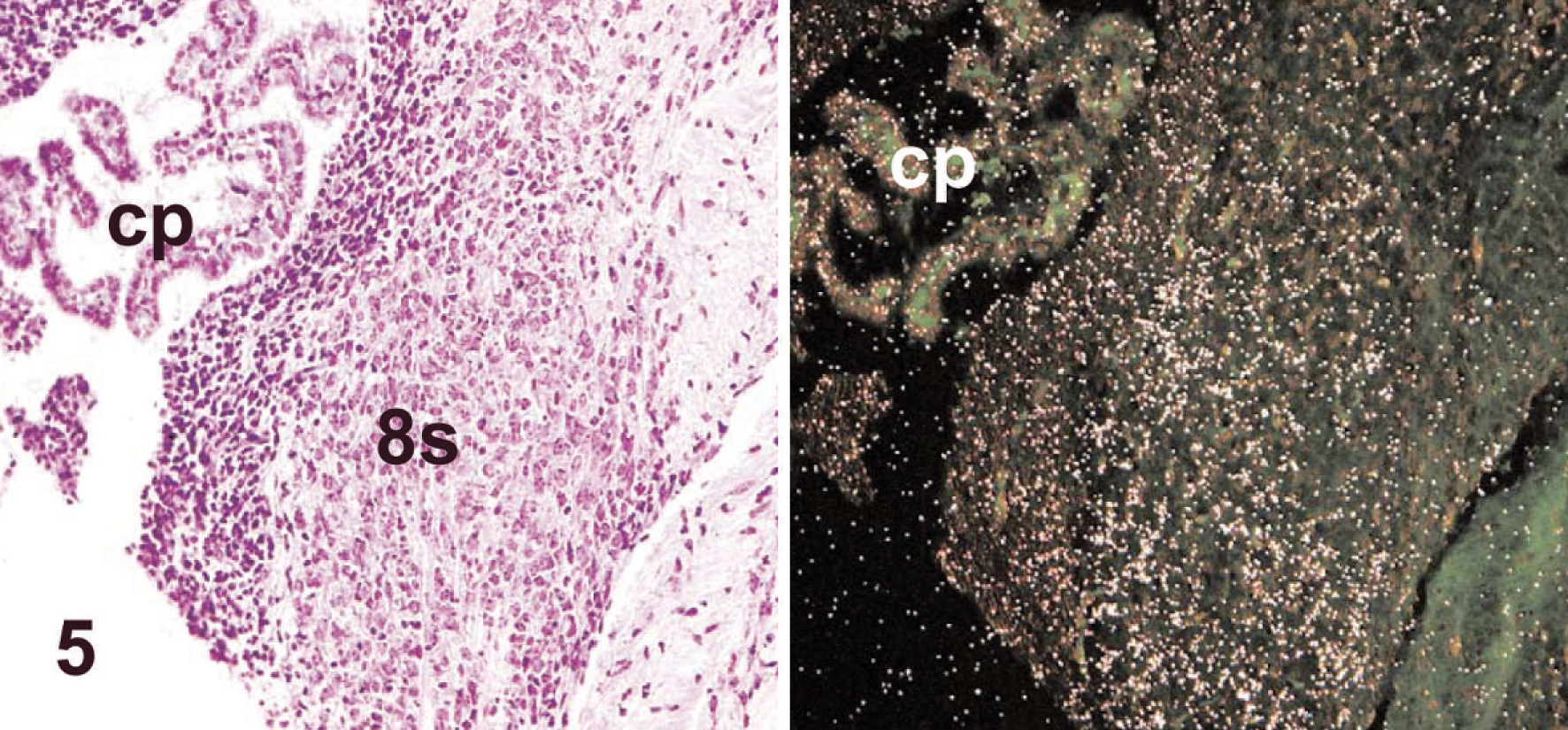
Brainstem; wild-type mouse neonate (postnatal day 1). Sensory neurons of the cochlear nucleus (8s) do express Clc diffusely in mild quantities. The choroid plexus of the fourth ventricle (cp) is identified for orientation. Stains: HE (left), isotopic in situ hybridization (anti-sense) for Clc (right).
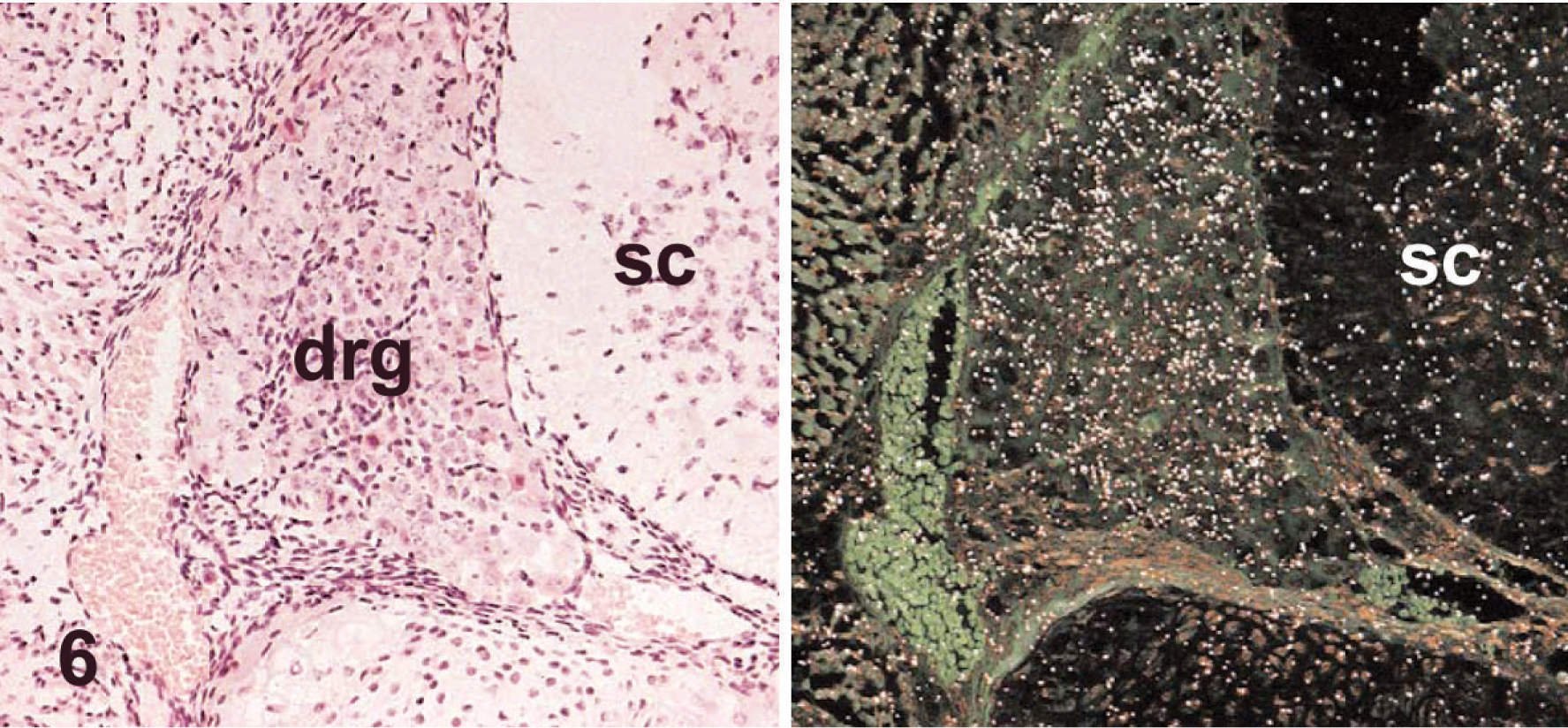
Dorsal root ganglion (drg), lumbar spinal cord; wild-type mouse neonate (postnatal day 1). Neurons exhibit diffuse, minimal Clc expression. The spinal cord (sc) is identified for orientation. Stains: HE (left), isotopic in situ hybridization (anti-sense) for Clc (right).
The recently discovered molecule CLC is a prominent neuropoietic cytokine. 2 Neuropoietic cytokines are important factors in regulating the survival and growth of neurons in developing and adult brain. To date, the only neuropoietic properties reported for CLC are its ability to sustain embryonic motor and sympathetic neurons in vitro 6, 8 and to promote astrocyte differentiation in neural stem cells. 10 The required participation of CLC in vitro 4 in a heterotrimeric signaling complex with CNTF-Rα and CLF strongly suggests that CLC serves the same roles in vivo. This hypothesis is supported by the neonatal lethal phenotypes and neuronal deficits that are observed in mouse pups lacking either Cntf-Rα 3 or Clf 5 and by the capacity for exogenous CLC to sustain developing neurons in ovo. 5 This experiment evaluated whether or not an equivalent phenotype (neonatal lethality because of site-specific neuronal deficits) also follows ablation of Clc.
As expected, Clc knockout mice had a reproducible phenotype (perinatal death, decreased facial motility, inability to suckle, and motor neuron deficits in the facial nucleus and ventral horn of the lumbar spinal cord; Table 2) that mirrored exactly the phenotypes of animals that lack either Cntf-Rα 3 or Clf. 1, 5 Thus, our current data in Clc null mutant mice—when considered together with prior reports that CLC along with CLF and CNTF-Rα are required components of a heterotrimeric signaling complex 4, 6 —provides strong evidence that CLC serves in vivo as one constituent of CNTF-II. Our data also suggest that CLC might also serve as a mediator for cell genesis in other neural regions, including dorsal root ganglia (Fig. 6), although any contribution appears to be confined to late (postnatal) developmental stages because neuronal deficits did not occur at this site in Clc knockout neonates (Table 2). This latter finding is interesting because neurons in dorsal root ganglia are also spared in mice lacking Cntf-Rα 3 or Clf, 1, 5 thereby suggesting that additional neuropoietic roles other than supporting cell survival remain to be elucidated for these molecules in other neuronal populations. An obvious example identified in this study is the cochlear nucleus, in which mild diffuse Clc expression is evident in neonates (Fig. 5). Further work with a conditional Clc null mutant will be necessary to explore the full scope of late-onset Clc expression and its functional relevance in this and other neuronal aggregates in the central and peripheral nervous systems.
Footnotes
Acknowledgements
We thank Mr. Tim Corbin and Dr. Venus Lai for assistance with the animal colony.