
Abstract
Mice harbouring a humanized liver represent a powerful tool for translating preclinical studies of drug metabolism and pharmacokinetics into humans, as well as the exploitation of basic studies on liver pathophysiology including hepatitis C virus (HCV) infection. Human adult stem cells injected into immunocompetent mice at preimmune stages of development, generate chimeric animals harbouring a liver with relatively discrete foci of human hepatocyte-like cells. In this study, we have evaluated whether similar protocol of xenotransplantation in the presence of selective pressure might lead to a higher human-into-mouse liver repopulation, leading to a relevant improvement of liver function. Human CD34+/CD133+ cells were microinjected into blastocysts from genetically-modified mice committed to develop a lethal hepatopathy, due to the absence of the enzyme fumarylacetoacetate hydrolase (FAH). Following xenotransplantation, mouse survival was followed over time and histochemical evidence of liver chimerism was assessed. The survival expectancy of seven out of 21 intrablastocyst xenotransplanted FAH knockout (Fah−/−) mice was significantly higher as compared with non-xenotransplanted mice. Several nodules of human hepatocyte-like cells were revealed by immunohistochemistry in the liver of rescued mice. Our data positively support the hypothesis that preimmune xenotransplantation of human stem cells into immunocompetent mice harbouring a lethal hepatic disease might lead to a functionally relevant human-mouse liver chimerism and marks a significant advancement towards the establishment of a novel translational preclinical model for liver diseases.
During the last decade, the generation of transgenic mice expressing human transgenes has proved to be a valuable tool toward achieving the humanization of mouse liver machinery. 1,2 In this respect, the most ambitious attempts are those aimed at the global humanization of the entire mouse liver including xenotransplantation of human hepatocytes into immunosuppressed mice with genetic liver disease. 3–5
However, despite the great success obtained using Alb-uPA/SCID/Bg and Fah−/−Rag2−/−Il2rg−/− mice, 4,5 a series of limitations hamper a more extensive use of these models, 6 such as the prerequisites of immuno-suppression, the availability of a regular source of fresh healthy hepatocytes, the occurrence of somatic silencing mutations and the need for skilful surgery in the first few days after birth.
Nonetheless, though insufficient studies characterizing liver function in such models, 7 many hopes are indeed envisioning a great value for human into mouse liver chimeras. Mice harbouring a humanized liver are expected to provide a better translation of preclinical studies of drug metabolism and pharmacokinetics into humans, as well as the exploitation of basic studies on liver pathophysiology including hepatitis C virus (HCV) infection.
The present report is an attempt to further explore alternative ways of humanizing mouse liver which in case of success might overcome most of previously mentioned limitations. 7 We have recently demonstrated that human adult stem cells injected into non-immunosuppressed mice at preimmune stages of development, even in the absence of selective pressure, can generate chimeric animals harbouring a liver with human hepatocyte-like cells. 8 Here, trying to go further with this approach, we propose similar procedures in mice affected by a pharmacologically controllable lethal hepatic disease such as tyrosinaemic mice. It was hypothesized that while developing tolerance to human antigens during the preimmune stage of development, selective pressure imposed by lethal murine hepathopathy might lead to a greater extent and/or improvement of chimeric liver function.
The absence of fumaryl-acetato-hydrolase (FAH) provokes the development of tyrosinaemia in both humans and animals. 9,10 Mice missing this gene die soon after birth, 11 unless appropriately transplanted with healthy hepatocytes or stem cells from healthy mouse donors, 12–15 or alternatively permanently treated with 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione (NTBC) capable of blocking the metabolism of tyrosine upstream. 16
In the present study, we investigated whether xeno-transplantation of human stem cells transplanted at the blastocyst stage from tyrosinaemic Fah−/− mice could lead to some improvement on liver metabolic functioning capable of rescuing or extending life expectancy.
Materials and methods
Animals
Fah−/− mice were originally generated in 1995 by M Grompe from Oregon Health and Science University (OHSU) University (Portland, OR, USA), 11 thereafter several colonies of Fah−/− mice were established by different laboratories. Fah−/− mice were generated from breeding pairs kindly provided by Dr Grompe to Dr Tripodi. All animal housing, handling and experimental procedures were carried out in accordance with national and company guidelines and the criteria outlined in the Guide for the Care and Use of Laboratory Animals. Moreover, animals were handled in absolute compliance with both European and Italian legislations (86/609 and DL116/92). Animals were housed in a controlled environment at a constant temperature (22 ± 1°C), relative humidity of 55±5%, 12 h light–dark cycle (lights on at 07:30 h) and HEPA filtrated air. Even if fully immunocompetent, Fah−/− colony and experimental animals were housed in specific pathogen free (SPF) conditions to avoid any potential pathogen contamination that could influence the observed phenotype. Animals received drinking water supplemented with NTBC at a dose of 1 mg/kg body weight per day. NTBC was purchased from Swedish Orphan Ltd (Stockholm, Sweden).
Intrablastocyst microinjection
Four-week-old female Fah−/− mice were injected with PMSG (5 IU/mouse) and two days later with HCG (5 IU/mouse) intraperitoneally and bred with adult male Fah−/− mice. Females were euthanized by CO2 inhalation at day 3.5 of gestation and murine blastocysts were isolated following standard procedures. Following a micromanipulator aided microinjection of 15–20 human cord blood CD34+/CD133+ (hCBCD34+/CD133+) or human fetal liver CD34+/CD133+ (hFLCD34+/CD133+) cells (Cambrex, East Rutherford, NJ, USA), blastocysts were incubated for 2–4 h in a CO2 incubator at 37°C and then transferred into CD1 foster mothers following commonly described procedures.
Histological studies
Mouse livers were collected, fixed in 10% formalin for 48 h and embedded in paraffin (bioplast special; melting point: 52–54°C). Five micrometre thick sections were adsorbed on polylysine-coated glass slides, air-dried, deparaffinized and rehydrated. Then, sections were stained with haematoxylin and eosin (H&E) for overall histopathological evaluation. Different sections were used for immunohistochemical studies. Before incubation with the primary antibodies, sections were unmasked by immersion into a preheated target retrieval solution (DAKO, Milan, Italy) for 15 min at 92–95°C. Endogenous peroxidase was blocked by incubating sections with 1% H2O2 in methanol for 30 min. Then, sections were incubated with the following human hepatocyte specific antibodies: monoclonal mouse anti-human haepatocyte paraffin 1 (HepPar1) (DAKO), monoclonal mouse anti-human cytokeratin-7 (hCK-7) (DAKO), rabbit anti-human alpha-1 antitrypsin (hAAT) (DAKO) and preadsorbed goat polyclonal anti-human serum albumin (HSA) (Bethyl Labs, Montgomery, TX, USA). HepPar1 and hCK-7 were labelled using DAKO ARK (Animal Research Kit, DAKO). Rabbit-anti-hAAT and goat-anti-HSA, after one hour of incubation at room temperature (RT), were detected with biotinylated anti-rabbit and anti-goat IgG, respectively. Sections were then incubated with streptavidin-peroxidase, ready-to-use (Labvision, Kalamazoo, MI, USA) for 15 min at RT. The reaction was developed by using diaminobenzidine (DAB) (0.075 g/L) and H2O2 (0.003%), for 5 min at RT. Following several washes with distilled water, slides were counterstained in haematoxylin, dehydrated and mounted in a non-aqueous permanent-mounting medium.
Enzyme-linked immunosorbent assay
Serum was harvested by retro-orbital bleeding. Human albumin in the murine blood was assessed by ELISA (Bethyl Labs) according to the manufacturer's manual. Serum dilutions of 10- to 10,000-fold were used to get values within the linear range of the standard curve.
Results
Human fetal liver CD34+/CD133+ or cord blood CD34+/CD133+ (hFLCD34+/CD133+, hCBCD34+/CD133+) cells were xenotransplanted by microinjection in Fah−/− blastocysts and reimplanted in CD1 foster mothers receiving drinking water supplemented with NTBC. Preimmune xenotransplantation of Fah−/− blastocysts with hCBCD34+/CD133+ and hFLCD34+/CD133+ cells resulted respectively in seven and 14 pups. Two days after birth, the dosage of NTBC in drinking water was reduced by half for one week, then further reduced for the following week to a quarter and finally a quarter dose of NTBC was transiently administered from 30 days postpartum for one week, alternated with a weekly drug-free interval, for three times. At the end of a six-week treatment, NTBC was definitively withdrawn. As shown in Figure 1, all control Fah−/− mice that were not xenotransplanted at the preimmune stage of development, died between days 10 and 40 postpartum (light blue line). Thirteen out of 14 Fah−/− mice xenotransplanted with hFLCD34+/CD133+ cells died during the first week after removal of NTBC, whereas only one mouse was rescued from death, surviving up to 133 days postpartum (orange line). Meanwhile, one out of seven Fah−/− mice xenotransplanted with hCBCD34+/CD133+ cells died one week after removal of NTBC, whereas the other six survived for several weeks (red line). The final survival profile was scored against age-matched non-transplanted Fah−/− mice (dark blue line). As clearly shown, whereas age-matched non-transplanted Fah−/− mice died during the first four weeks after removal of NTBC, Fah−/− mice receiving intrablastocyst xenotransplantation with hCBCD34+/CD133+ cells survived up to three times longer.
Time curve survival profiling. A total of 22 non-transplanted Fah−/− mice (light blue line) as well as seven and 14 Fah−/− mice receiving respectively intrablastocyst xenotransplantation with cord blood (red line) or fetal liver (orange line). CD34+/CD133+ cells were monitored after birth. In all cases animals received full dose NTBC during pregnancy, at birth reduced to half dose for the first week (dark green bar) and later reduced again to a quarter dose in the second week (light green bar), to be finally completely removed. In the case of Fah−/− mice receiving cord blood derived CD34+/CD133+ cells and surviving for up to 30 days after birth, three additional cycles of a quarter dose of NTBC were administered at weekly intervals (light green bars), but thereafter definitively removed. For comparison, age-matched non-transplanted Fah−/− mice receiving full dose NTBC (dark blue line), were subjected to NTBC removal (see online version for colour references)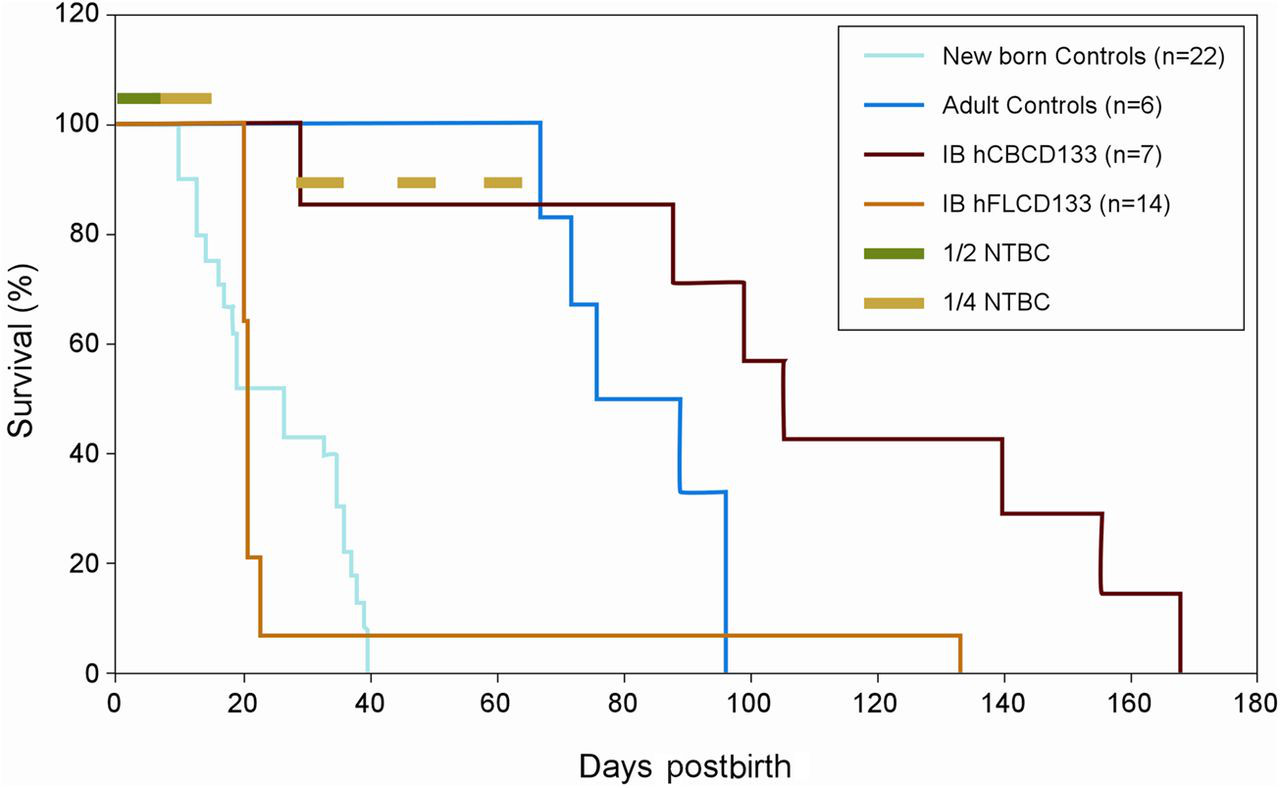
Trying to monitor in vivo the progression of chimerism, the presence of human albumin (hAlb) in the sera of intrablastocyst-xenograft-Fah−/− mice (IB-XT-Fah−/−) was periodically evaluated at two-week intervals after weaning by ELISA. However, neither hAlb nor hAAT was ever observed in the sera of IB-XT-Fah−/− mice, except for one animal transplanted with hFLCD34+/CD133+ cells that showed significant levels of hAlb (50 µg/mL) at postnatal day 120. Though limited, this represents further evidence confirming a chance of relatively large fully functional human engraftment (2%) into chimeric mice.
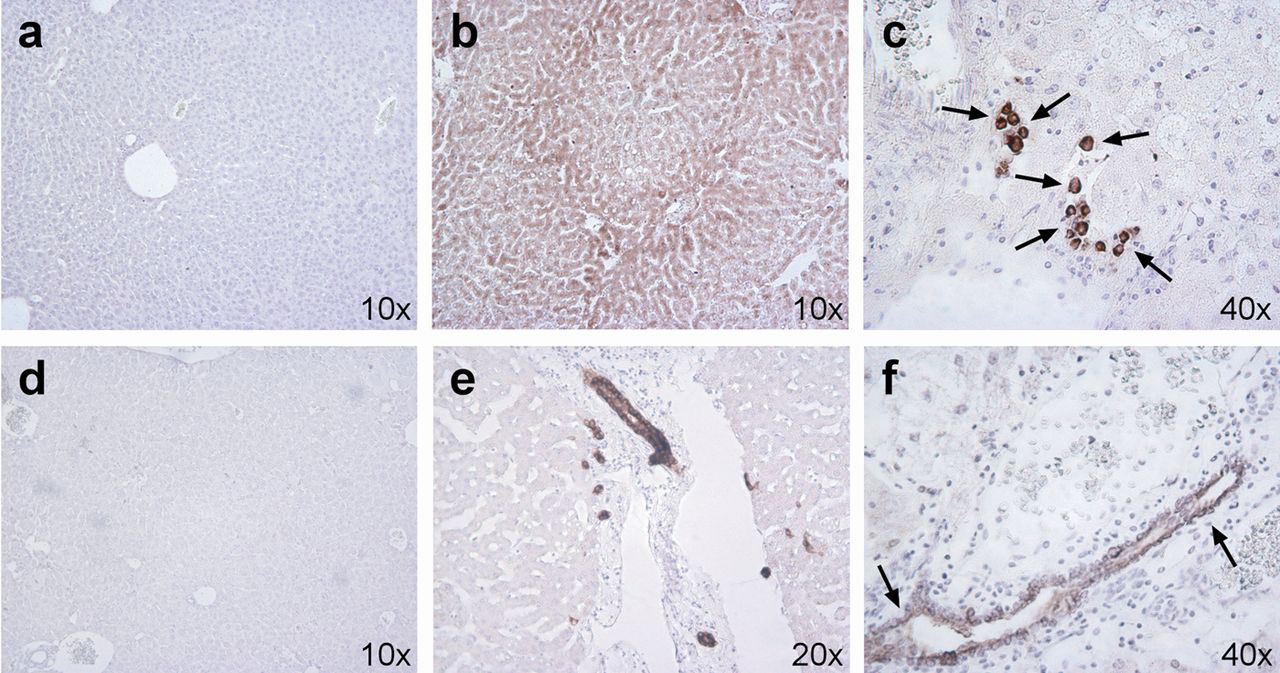
At death, the livers and kidneys from all IB-XT-Fah−/− mice were removed and macroscopically examined and compared with non-transplanted Fah−/−. Interestingly, the livers from IB-XT-Fah−/− mice showed the presence of several nodules of apparently surviving tissue. Swollen and pale kidneys were macroscopically detected in most IB-XT-Fah−/− mice, which was not obvious even in Fah−/− mice following NTBC removal (Supplementary Figure S1, see Appearance of liver and kidneys following haematoxylin/eosin staining. Obvious signs of very extended and severe hepatic damage manifested in non-transplanted Fah−/− mice that died upon removal of NTBC (b), were contrasted against Fah−/− mice sacrificed under NTBC treatment (a) in which no evidence of injury was observed, as well as chimeric Fah−/− mice that died several weeks after NTBC withdrawal (c), which in the middle of relative large injured zone exhibited importantly huge nodular areas of surviving tissue. By contrast, chimeric animals exhibited very strong evidence of nephropathy (f), not observed in Fah−/− mice sacrificed under NTBC treatment (d) or Fah−/− mice that died upon removal of NTBC (e) Identification of human hepatocyte-like cells in liver of chimeric mice upon immunostaining to hepatocyte-specific antigen. Supporting the specificity of the antibody, it is possible to observe staining of human liver section (a) which is not observed in a representative liver section coming from a negative control non-transplanted Fah−/− mouse that died upon NTBC withdrawal (b) or from chimeric mice when omitting incubation with the primary antibody (d). By contrast, most IB-XT-Fah−/− chimeric mice exhibited relative large nodular areas harbouring cells that stained positive for hepatocyte-specific antigen (c), which can also be observed at higher magnification (e) Identification of two additional human liver specific markers in the liver of chimeric mice. Specificity of the immunohistochemical techniques used for human alpha-1 antitrypsin and hCK-7 is respectively provided by confronting positive staining observed in human liver sections (b, e) and absence of staining in negative control liver samples coming from non-transplanted Fah−/− mice dead upon removal of NTBC (a, d). Presence of discrete small foci of round cells positive for human alpha-1 antitrypsin was observed in chimeric mice (c). Similarly, several foci of hCK-7 positive cells, organized in structures resembling bile ducts were observed in some chimeric mice (f)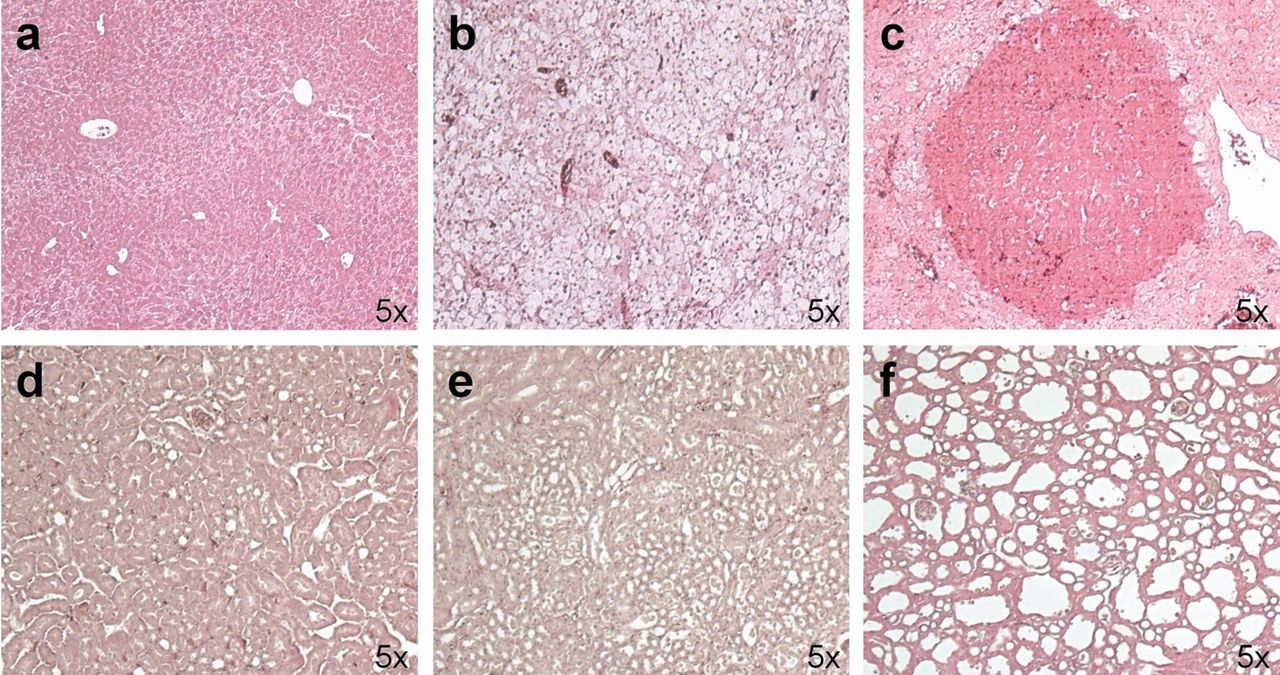
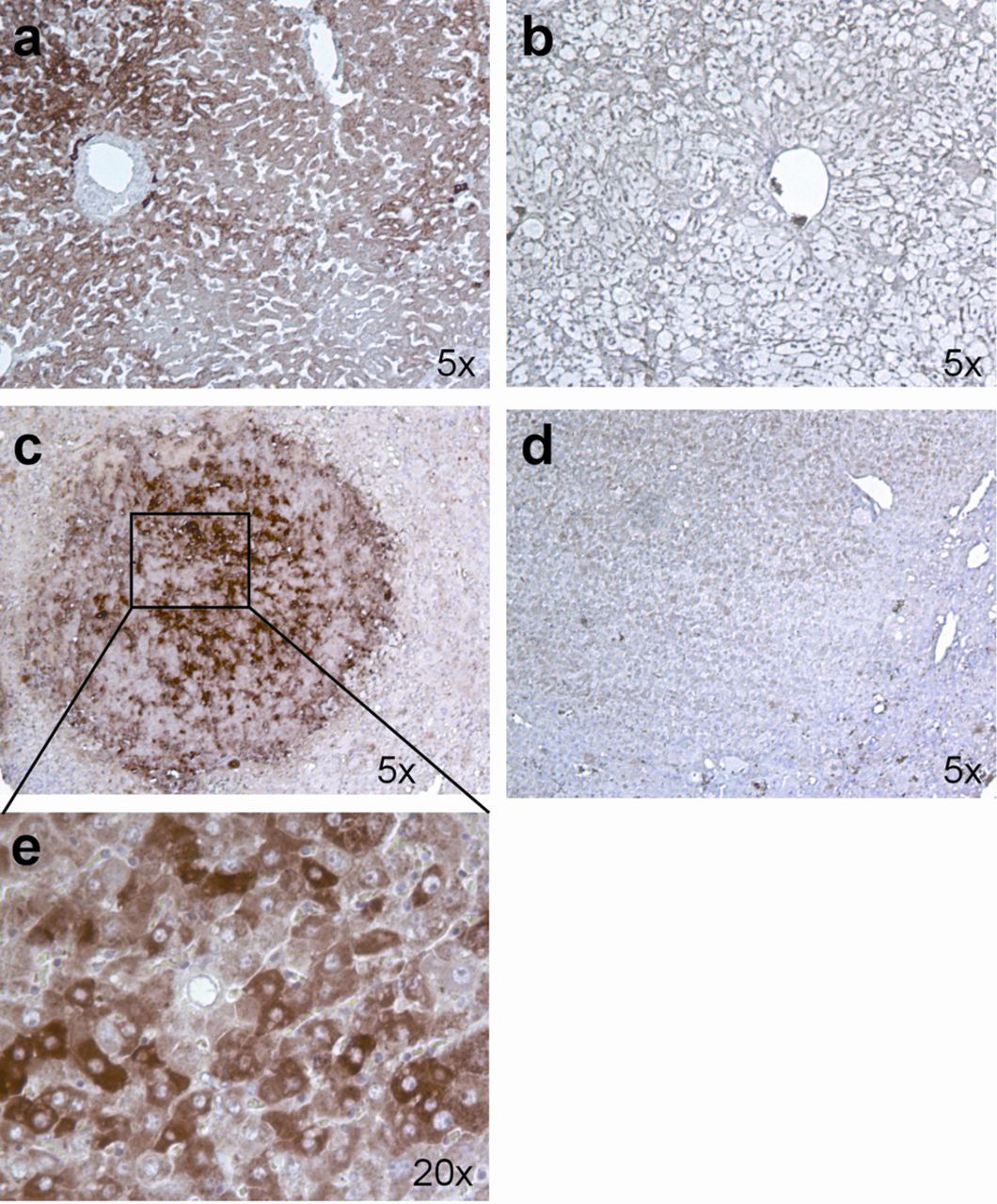
To provide a semiquantitative idea of the extension of the engraftment we observed that Hepar1 staining was extended through well-defined nodules of surviving cells (reaching 5–15% of the staining area in the whole liver cross-sections), while AAT staining was much more confined to reduced foci in the perivascular regions (reaching less than 1% of the staining in whole liver cross-sectional areas). Similarly CK7 staining, as compared with HepPar1, reached a much modest extension (<1%) confined to the neighbouring biliary duct-like structures.
Discussion
Fah−/− mice undergo severe liver damage as a consequence of the genetic knockout of fumarylacetoacetate hydrolase (Fah) gene, which plays a key role in the catabolism of tyrosine. 10 Absence of the FAH enzyme causes the accumulation of fumarylacetoacetate (FAA) which is an extremely reactive and toxic metabolite resulting primarily in a lethal hepatopathy. 9 In normal mice, expression of the FAH enzyme starts at day 16 of fetal development, 17 but the dramatic effect of FAH deficiency becomes evident within the first 12 h after birth when homozygous animals die due to severe hypoglycaemia and liver dysfunction. 11 The survival of these knockout mice is made possible by prenatal and lifelong chronic treatment with NTBC, a drug capable of blocking upstream the metabolic pathway of tyrosine and therefore avoiding the accumulation of the hepatotoxic intermediate. 16 We have previously demonstrated that, even in the absence of any selective advantage, injecting human cord blood derived CD34+ cells during the preimmune stage of mouse development can result in the generation of tolerant chimeras harbouring a liver with discrete foci of hepatocyte-like cells. 8 Taking this work one stage further, here we illustrate that non-embryonic human stem cells are able to more extensively repopulate the liver with a relevant increase in the lifespan of immunocompetent Fah−/− mice, when injected at the blastocyst stage of development. To this purpose, in the search for a more ancestral subpopulation of stem cells, we decided to use CD34+/CD133+ cells from both human umbilical cord blood and fetal liver. As previously reported, this subpopulation of cells might not only behave as primitive progenitor cells for the haematopoietic system, 18 but also demonstrate a pluripotent capacity to repopulate non-haematopoietic tissues such as the liver. 19 Indeed, by xenotransplanting these cells into blastocysts from Fah−/− mice, upon removal of NTBC we were able to observe a considerable increase in life expectancy as compared with non-transplanted Fah−/− mice, indirectly indicating a transient or partial metabolic rescue of Fah−/− mice. Supporting the idea that the engraftment and expansion of human stem cells is the basis of the observed increase in life expectancy, we observed the presence of large areas of immunostaining for a human liver specific marker such as HepPAR1 and hAAT in the IB-XT-Fah−/− mice. It is worth noting that hAAT immunostaining was not as extensive and evident as HepPAR1, consistent with our failure to detect any hAAT in sera, probably indicating an insufficient differentiation stage of human stem cells. On the other hand the identification of hCK-7-positive cells organized in structures akin to human bile ducts, argues in favour of an ongoing overall repopulation of the mouse liver by human cells.
A critical issue which deserves further investigation is whether the observed chimerism was actually due to stem cell differentiation or stem cell fusion. There is much contrasting evidence in the literature arguing in favour of stem cell transdifferentiation and/or stem cell fusion potential. 20 Preliminary studies using the Fah−/− system suggest a prominent/notable differentiation potential for different mouse adult stem cell subpopulations, 14 but at the same time other subsequent studies have demonstrated that most of the chimerism observed was actually due to stem cell fusion capacity. 21,22 By contrast, Azuma et al. 5 demonstrated the lack of cell fusion in Fah−/− mice. Nonetheless, independently of which major pathway it goes through, an indisputable form of human-into-mouse liver chimerism was obtained. It remains unclear, however, why we obtained restricted liver humanization with just a partial improvement in life expectancy, instead of total liver repopulation providing definitive protection against death. In this regard, it might be worth investigating whether CD34+/CD133+ cells might be ancestral enough to achieve complete hepatocyte differentiation, since we failed to detect major late markers of mature hepatocytes such as hAlb. Moreover, it is known that ancestral stem cells are capable of displaying infinite proliferative potential, whereas the more committed stem cell subpopulations exhibit a more limited duplication number, 23 which in our case might explain incomplete liver repopulation.
Alternatively, it cannot be excluded that the death of our chimeric animals was due to collateral renal failure rather than an insufficient recovery of the liver. In other words, the subpopulation of stem cells we used might be good enough to correct liver dysfunction, but not to overcome the concomitant kidney disease. In fact, our chimeric animals exhibited dramatic signs of severe renal disease, consistent with a previous report
24
(see Figure 2 and Supplementary Figure S1, see
Undoubtedly there are still many open issues to resolve and the efficiency of this novel approach to generating chimeric mice is limited. Nonetheless current data positively support further exploration of this original strategy to generate humanized mice. There has been previous evidence in the literature suggesting that metabolic dysfunctions can be corrected by transplanting stem cells at the preimmune stage of development. Transplantation in utero with fetal liver cells has been reported to alleviate many effects of lysosomal stromal disease in mice. 26 Likewise, haematopoietic stem cells transplanted in utero were effective in producing muscle chimerism in mdx mice affected by muscular dystrophy. 27
In summary, we found that human CD34+/CD133+ cells microinjected into blastocysts from Fah−/− mice increases their survival repopulating the damaged murine liver with human hepatocyte-like cells. The present study, in parallel to outlining the value of human CD34+/CD133+ cells for the generation of immunocompetent humanized mice, might also provide further evidence in favour of the therapeutic potential of human haematopoietic stem cells. The use of alternative subpopulations of human stem cells, and/or diverse strains of hepatically compromised mice, or varying dosing schedules of NTBC to achieve an appropriate rate of selective advantage, might be considered in the future to further optimize this novel approach.
In conclusion, we believe that the preimmune stage transplantation approach for the creation of mice with humanized liver is promising because (i) it offers a possibility of avoiding the need of immunosuppression in the mice and (ii) to achieve this goal it is possible to apply the well-known and widespread blastocyst injection technology. Although further studies are needed, this new approach holds the promise of becoming the method of choice for creating mouse models with humanized liver.
Footnotes
ACKNOWLEDGEMENTS
This work was supported in part by grant AIRC IG 10507 to L Aurisicchio. We thank Dr Markus Grompe for providing Fah−/− mice, Romina Sasso and Fabrizio Colaceci for technical support; Janet Clench and Rosa Angela Colamarino for language editing.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.