
Abstract
Mouse models with targeted disruptions of coagulation factor genes are used to study disorders of haemostasis such as haemophilia. Standard protocols to obtain biopsies for genotyping in breeding programmes are based on invasive sampling methods such as tail clipping. These procedures imply a high risk of fatal bleeding, especially in haemophilic mouse models. Here we used a non-invasive sampling method obtaining stool samples for DNA isolation in a breeding programme, aiming to introduce targeted disruptions of Fc receptor genes (Fc gamma receptor IIB and III) into the haemophilia A mouse model (factor VIII deficiency). Faecal pellets were reliably obtained from individual mice and high-quality DNA was extracted with a mean yield of 7.1 μg/pellet. Polymerase chain reaction amplification of wild-type and knockout alleles for Fc receptor and factor VIII genes was similar, comparing stool and peripheral blood as the source of genomic DNA. Definite genotype identification was achieved in a first attempt in 336 of 352 analyses (95%). Repeated analysis of homozygous knockout animals confirmed the first result in all cases. No animal was lost due to bleeding from the procedure. In conclusion, DNA isolation from stool is a preferable method for genotyping in laboratory animals, especially in models of bleeding disorders. Avoiding loss of animals due to bleeding implies a substantial improvement in animal welfare by reducing the number of animals used and may also advance the effectiveness of breeding programmes in these disease models.
Mouse strains with targeted disruption of coagulation factors are important research tools to investigate the pathogenic mechanisms and treatments of bleeding disorders such as haemophilia or von Willebrand syndrome. Resembling the human disorders, murine haemophilia A and B are X chromosome-linked recessive traits. Homozygous females and hemizygous males have a factor VIII (FVIII) or IX activity of <1 IU/dL (<1% of normal) and exhibit prolonged clotting times. 1,2 These models are particularly useful to study the formation of anti-FVIII inhibitory antibodies that are nowadays the most serious complication of replacement therapy in haemophilia. The potential role of immunomodulatory genes for the development of inhibitory antibodies is of major interest in this area. Crossbreeding of the haemophilic mouse model to strains carrying disruptions of immune response modifiers is a promising approach to address this issue.
Mice with a targeted disruption of the FVIII gene are viable and fertile. No bleeding difficulties are apparent during birth. However, severe bleeding can result from routine procedures such as ear tagging, tail clipping or blood withdrawal. These difficulties establish significant problems for breeding programmes requiring frequent genotype identification. Conventional protocols for DNA isolation are still based on tissue biopsies from the toe, ear or tail. These methods not only exert significant stress, pain and mutilation, but also imply a high risk of exsanguination in mouse models of bleeding disorders.
Extraction of genomic DNA has also been reported from faecal pellets. 3 These are easy to obtain using non-invasive methods and provide an excellent source of genomic DNA, given that murine intestinal epithelia regenerate every three to four days and old cells are delivered into the lumen. Here, we report on the use of this method in a large haemophilic mouse model breeding programme. Results of genotyping using DNA from stool were confirmed using DNA obtained from peripheral blood demonstrating reliability of this method.
Materials and methods
Mouse strains and breeding
All experimental procedures were performed in accordance with the accepted principles of welfare of animals used in science and were approved by the regulatory authority. The X-linked haemophilia A mouse model used in this study (F8: B6;129S4-F8 tm2Kaz ) was generated by targeted disruption of exon 17 using a neomycin resistance gene. 1 Haemophilic females were mated to males with homozygous targeted disruptions of autosomal immune response modifying genes of interest, specifically the Fc gamma receptor IIb (FcgR2b: B6;129S4-Fcgr2b tm1Ttk /J) 4 and Fc gamma receptor III (FcgR3: B6;129P2-Fcgr3 tm1Sjv/J). 5 It can be assumed that all F1 offspring are heterozygous for the Fc receptor gene disruption and hemizygous (males) or heterozygous (females) for the FVIII gene disruption. In the next generation, F2 offspring were screened for homozygous Fc receptor gene disruption and hemizygous (males) or homozygous (females) FVIII gene disruption.
Sampling
Stool samples were obtained by taking individual mice out of their cage and collecting a faecal pellet in a 2 mL reaction tube. More than 90% of mice excreted a faecal pellet within 1 min. Blood samples obtained for confirmation of results were taken by retro-orbital puncture.
DNA isolation
Genomic DNA from faeces was isolated using a QIAamp DNA stool mini kit (Qiagen, Hilden, Germany). Briefly, a faecal pellet was homogenized and lysed in 1.6 mL ASL buffer. Following centrifugation, InhibitEX tablet was added to adsorb impurities that can degrade DNA and inhibit downstream reactions. After centrifugation, the supernatant was digested with proteinase K and DNA was precipitated with ethanol. DNA was further washed and purified by a QIAamp spin column according to the manufacturer's instructions and finally eluted in 200 μL AE buffer. The DNA concentration was determined by UV spectrophotometry of a 1:1000 dilution using the formula:
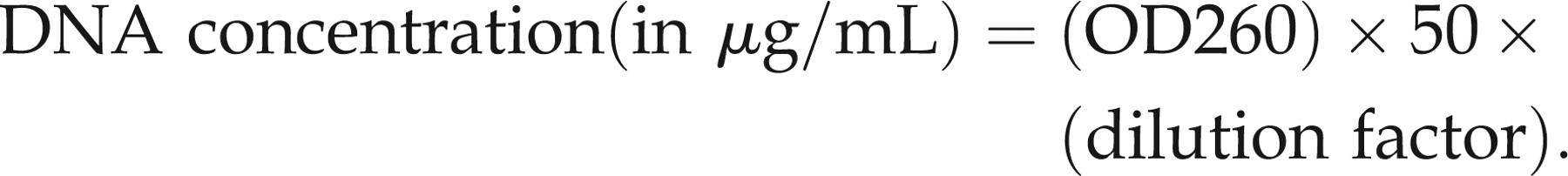
Genomic DNA from peripheral blood was isolated using DNA isolation kit for mammalian blood (Roche, Mannheim, Germany) according to the manufacturer's instructions.
Polymerase chain reaction
Three primers were used to detect each allele as given in Table 1, i.e. a specific primer for wild-type and mutant allele and a common antisense primer. For detection of the FcgR2b alleles, a duplex polymerase chain reaction (PCR) was set up containing all three primers in one sample. For F8 and FcgR3, wild-type and disrupted alleles were detected separately. PCR was done in a final volume of 50 μL using Hotstart Taq Master Mix Kit (Qiagen, Hilden, Germany). The mixture contained 6 pmol of each primer, 5 ng of template DNA and 25 μL Hotstart Taq Master Mix (with reaction buffer, 0.5 μmol/L deoxyribonucleotide triphosphate, 2.5 mmol/L MgCl2 and 2.5 U of Hotstart Taq DNA polymerase). PCR was performed using a thermal cycler (T3 thermal cycler, Biometra, Göttingen, Germany) with the following cycling programmes: for F8, one cycle at 95°C for 15 min, followed by 34 cycles at 94°C for 30 s, 61°C for 30 s and 72°C for 45 s, and final extension at 72°C for 10 min; for FcgR2b and FcgR3, 95°C for 15 min, followed by 34 cycles at 94°C for 30 s, 62°C for 45 s and 72°C for 45 s, and final extension at 72°C for 10 min. PCR containing only reagents without sample DNA was run as a negative PCR control. PCR products (5 μL) were evaluated on 1.5% (w/v) agarose gel in 1× TBE buffer (89 mmol/L Tris, 89 mmol/L boric acid, 2.5 mmol/L ethylenediaminetetraacetic acid, pH 8.3) containing ethidium bromide.
Polymerase chain reaction primers
Results
DNA extraction from stool yielded an average of 7.1 μg/pellet (range 0.1–50 μg). Purity of the DNA preparation as indicated by the OD260/OD280 ratio was 1.5 (range 0.3–2.9). To validate the detection of wild-type and knockout alleles, DNA samples from stool and peripheral blood were analysed in parallel. Amplification of the F8, FcgR2b and FcgR3 alleles from either knockout or wild-type animals resulted in strong and reproducible signals (Figure 1). Comparing blood and stool as the source of PCR template, there was no relevant difference with respect to the amount or purity of the PCR product. Regarding FcgR2b detection, wild-type and knockout alleles could be easily analysed in a single duplex PCR assay.
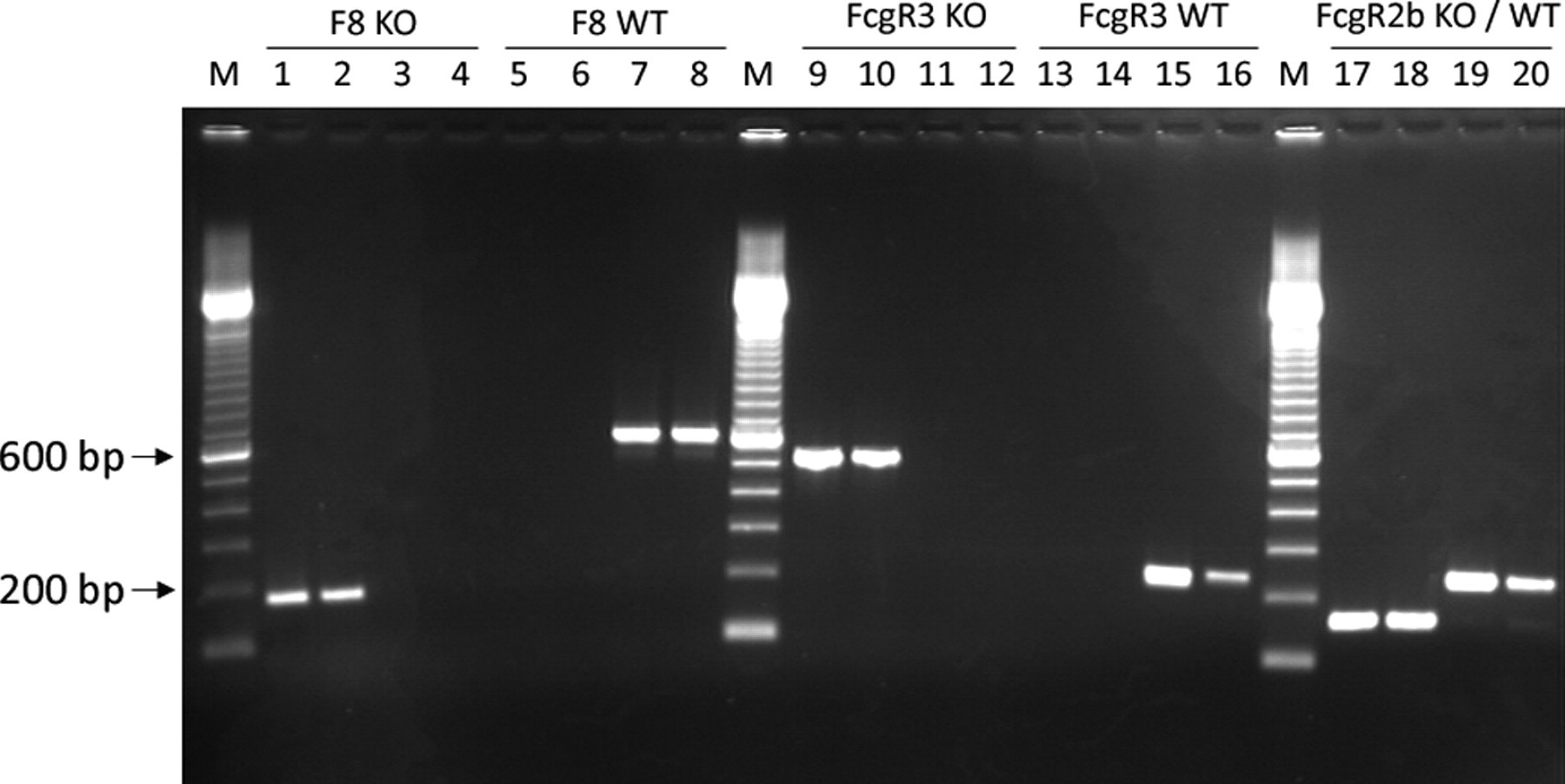
Detection of knockout and wild-type alleles from blood and stool samples. PCR products amplified from stool (even lane numbers) compared with peripheral blood (uneven lanes). Detection of the F8 knockout (KO) alleles (lanes 1–4) and F8 wild-type (WT) alleles (lanes 5–8) in F8−/− mice (lanes 1, 2, 5, 6) and WT mice (lanes 3, 4, 7, 8). Detection of FcgR3 KO alleles (lanes 9–12) and FcgR3 WT alleles (lanes 13–16) in FcgR3−/− mice (lanes 9, 10, 13, 14) and WT mice (lanes 11, 12, 15, 16). Simultaneous duplex PCR detection of FcgR2b KO/WT alleles (lanes 17–20) in WT mice (lanes 17, 18) and FcgR2b−/− mice (lanes 19, 20). M: molecular weight marker
PCR analysis of samples was used for genotyping in a breeding programme aiming to introduce the FcgR2b and FcgR3 knockout alleles into the F8−/− mouse model. Samples were obtained from four-week-old mice. The genotype could be identified in a first attempt in 336 (95%) of a total of 352 analyses performed (Table 2). The genotype of double knockout mice considered for further breeding was confirmed from a second sample with no conflicting results obtained.
Screening results of genotyping from murine stool samples
Discussion
Faecal pellets can be used as a source of high-quality genomic DNA in mice. 3 Here, we report for the first time on the use of this method in a large breeding programme. Our results demonstrate that DNA purified from stool using a simple and fast method was a reliable template for routine genotyping of mice in a large breeding programme. The success rate, as defined by unambiguous genotype identification in a first attempt, was 95%. Duplex PCR detection of more than one allele is possible using this method.
During the entire programme, no mice were lost due to bleeding from invasive sampling. In contrast, approximately 10–20% of haemophilic mice are lost following blood withdrawal by retro-orbital puncture. 6,7 Tail clipping as routinely used to obtain DNA samples for genotyping is also associated with a risk of death of up to 90% due to excessive blood loss. 6,8 Cauterization with silver nitrate can improve survival rates after tail clipping, but results in stress, discomfort and pain for the animal and is often not successful. Connelly et al. 9 reported a loss of approximately 30% of haemophilic mice using cauterization of tail clips. Ear biopsies are probably less hazardous in these mice, but have also been associated with profuse bleeding occasionally (ibid).
Tail clipping implies additional problems apart from bleeding. Some authorities suggest the use of anaesthesia to reduce stress and pain, which is discussed controversially. In a recent comparative study, tail biopsy during anaesthesia with diethylether or methoxyflurane induced long-lived alterations in heart rate, body temperature and body weight in comparison with tail biopsy without anaesthesia. Anaesthesia using diethylether also resulted in a mortality rate of 7%. 10 According to European and North American regulations, the choice of procedures should aim to minimize pain, suffering and distress for the animal, especially if alternative methods are available. 11 Concerning the choice of tissue for DNA sampling, non-invasive sampling methods such as stool collection should be considered less harmful than tail clipping.
In addition to stool samples, alternative sources for genomic DNA may be saliva, epithelial cells from the cheek or rectum, or hair bulbs. Five microlitres of saliva from adult mice were sufficient for genotyping by standard PCR amplification in transgenic mice. 12 However, saliva collection in young mice required flushing off the oral cavity with sterile water using a plastic pipette and a nested PCR for reliable genotyping results (ibid). Isolation of buccal endothelial cells by scraping the inner cheek with a scoop was successful for genotyping for at least 50 PCRs from newborn mice. However, this method was technically demanding and required a special oral speculum providing a soft pressure to open the mouth. 13 Epithelial cells from the rectum were also obtained by scraping off the inner surface of the rectum with a plastic inoculation loop, which was effective to analyse three different lines of transgenic mice by standard PCR. 14 This method also requires special operating skills and may cause considerable handling stress for the animals. Hair bulbs were used for DNA detection by PCR after alkaline lysis in more than 100 animals and proved to be a simple and fast procedure. 15 A potential limitation of this method is the risk of cross-contamination and false-positive results, especially when large numbers of mice are screened. Stool samples are easy to obtain without excessive distress, pain or injury to animals. Our results demonstrate that this method can be reliably applied in a large-scale breeding programme.
In summary, stool samples are a preferable source for DNA isolation. This non-invasive method reduces discomfort and stress to the laboratory animal and reduces animal loss due to excessive bleeding. Moreover, the method provides a high-quality source for genomic DNA; it is simple, fast and inexpensive to perform and can be repeated without limitations. We therefore suggest using stool samples for genotyping of laboratory animals in breeding programmes, especially mice with bleeding disorders.