
Abstract
Summary
The expression and immune modulation of Epstein-Barr virus-encoded oncogene latent membrane protein 1 (N-LMP1) is essential in the pathogenesis of nasopharyngeal carcinoma. In previous studies, cell transformation has been induced by the expression of EBV-encoded N-LMP1 in non-tumour BALB/c-3T3 cells and these cells have then been used to form tumours in T-cell-deficient nude mice. However, studies using this model have been limited by the lack of a competent immune system. To facilitate the study of immune components in N-LMP1-driven oncogenesis, we herein developed a simplified N-LMP1-derived tumour model in immunocompetent mice. Cell transformation was induced by the expression of N-LMP1 in BALB/c-3T3 cells, and these transformants were used to induce oncogenesis in BALB/c mice. In contrast to the 100% successful tumour-induction rate in nude mice treated with monodispersed transformed cells, the tumour incidence in BALB/c mice was only 5–36%. However, the transplantation of tumour fragments into BALB/c mice yielded a reproducible tumour-induction rate of >85%, which is acceptable for most of the research needs. This novel model of N-LMP1-directed oncogenesis in an immunocompetent environment may serve as an important platform for the future assessment of N-LMP1-targeted tumour therapies.
Keywords
Nasopharyngeal carcinoma (NPC) is a retro-nasal malignant tumour consistently associated with the latent state of Epstein-Barr virus (EBV) infection. The expression of EBV-encoded oncogene latent membrane protein 1 (LMP1) is considered as a determining factor for the processes of cell immortalization and malignant transformation (Wang et al. 1985, Baichwal & Sugden 1988, Raab-Traub 2002). LMP1, a member of the tumour necrosis factor receptor family, possesses the unique property of exerting both oncogenicity and immune-modulatory functions by ligand-independent activation of multiple intracellular signalling pathways and downstream genes (Gires et al. 1997, Li & Chang 2003). Protein sequence analysis has identified two major types of LMP1: prototype LMP1, originally identified in EBV-positive B lymphoma (B95-8) cells (B-LMP1) (Kaye et al. 1993); and N-LMP1, a variant with multiple mutations and a 10-amino acid deletion at the C-terminus, isolated from NPC endemic areas, including Southern China and Taiwan (Hu et al. 1991, Chen et al. 1992, Busson et al. 2004). In EBV carriers, mixed B-LMP1 and N-LMP1 infections are frequently observed. However, N-LMP1 is the prevalent Asian NPC variant (Hu et al. 1991, Chen et al. 1992, Lin et al. 2001, Li & Chang 2003, Edwards et al. 2004), implying that N-LMP1 might have a selection advantage over B-LMP1 in NPC carcinogenesis among individuals in this part of the world.
The cytotoxic T lymphocyte (CTL) response is the most important immune control directed against latent state viral gene products. Studies have identified the major histocompatibility complex class I (MHC I)-restricted B-LMP1 epitopes recognized by CTLs in healthy, EBV-seropositive individuals (Khanna et al. 1998). In contrast, researchers have been unable to identify the CTL epitopes for the N-LMP1 variant in NPC samples from Chinese individuals (Burrows et al. 2004, Edwards et al. 2004, Lin et al. 2004), suggesting that N-LMP1 may evade the immune response, thus playing a crucial role in the development of NPC in this population. Indeed, the presence of N-LMP1 was recently identified as a molecular marker for NPC diagnosis in Taiwan (Lin et al. 2001). LMP-1 expression in NPC specimens has been linked to local production of chemokines, which attract the leukocyte migration required for stromal formation (Niedobitek et al. 1996, Yoshizaki et al. 2001). Since leukocyte stromal development is a prerequisite for NPC oncogenesis (Niedobitek et al. 1996), researchers are currently seeking to understand how N-LMP1 directs the communication between preneoplastic epithelial cells of the nasopharynx and the infiltrating leukocytes (e.g. macrophages, B-cells and T-cells). In addition, it is important that we examine whether N-LMP1 tumours can be controlled by the immune system, and whether N-LMP1 can be targeted for therapeutic intervention under normal immune conditions. These and other studies require appropriate animal models of N-LMP1-induced tumours.
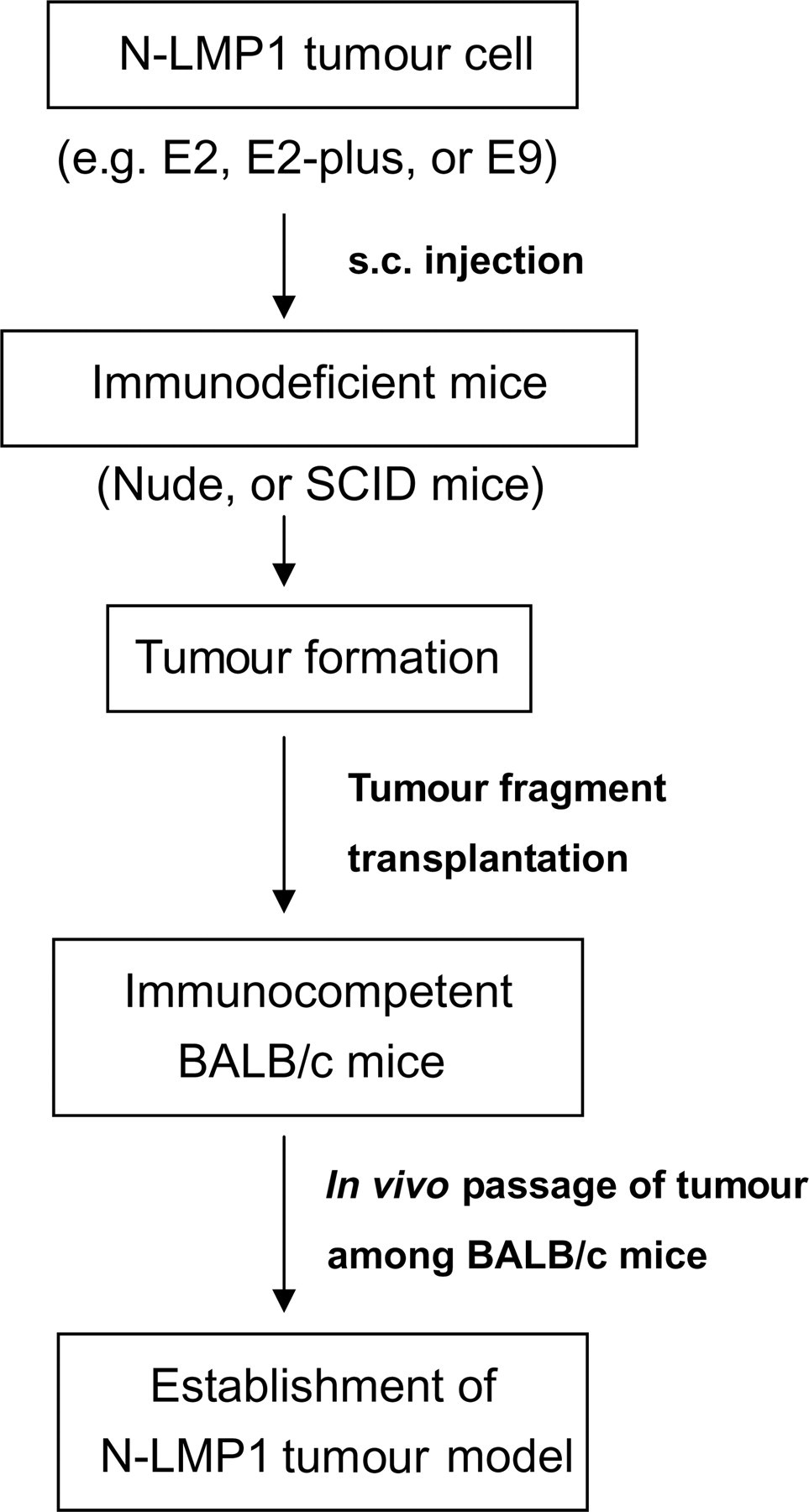
In particular, N-LMP1 tumour mouse models in immunocompetent hosts can serve as a crucial in vivo system for investigating the role of immunecompetency and assessing specific therapeutic strategies against N-LMP1-directed oncogenesis. In previous studies, a subcutaneous (s.c.) injection of tumour cell line-derived LMP1-expressing transfectants has been used to induce LMP1-expressing tumours in immunocompetent mice (Trivedi et al. 1994, Hu et al. 2000). However, the use of this approach has been limited by complications in separating the LMP1-associated changes from those associated with the tumour cell line background. Other studies have focused on the observation that N-LMP1 is the only known EBV gene capable of transforming BALB/c-3T3 cells, which have a non-tumour background (Wang et al. 1985, Baichwal & Sugden 1988). The transfection of N-LMP1 into BALB/c-3T3 cells generates a simplified system in which researchers have been able to examine the transforming effects specifically mediated by N-LMP1. In addition, s.c. injection of N-LMP1-transfected BALB/c-3T3 cells (3T3/N-LMP1) successfully induces tumour formation in T-cell-deficient nude mice (Chen et al. 1992, Li et al. 1996, Yu et al. 2002). However, the induction of N-LMP1 tumours in nude mice does not mimic the pathological situation in the normal host. In order to investigate the immune mechanisms in N-LMP1-directed oncogenesis and specific therapeutic strategies against this cancer, researchers need a simplified in vivo N-LMP1 tumour model in an immunocompetent host.
We herein sought to develop such a model. Since BALB/c-3T3 cells are theoretically syngeneic to BALB/c mice, we tested the use of 3T3/N-LMP1 cells to establish an N-LMP1 tumour model in normal BALB/c mice. We found that monodispersed tumour cells induced tumours at a very low rate in BALB/c mice. However, additional experiments revealed that transplantation of tumour fragments yielded an acceptably high tumour-take frequency (>85%), indicating that this system could be used to generate a simplified N-LMP1-directed tumour model in an immunocompetent host. As the N-LMP1 oncoprotein is the major driving force for cellular transformation, this novel model system may facilitate studies of N-LMP1-related immune modulation, as well as serving as a basis for evaluating the therapeutic efficacy of candidate agents and/or immunological manipulations.
Materials and methods
Mice and housing conditions
Male BALB/cByJNarl mice (6–10 weeks, 20–30 g) were purchased from the National Laboratory Animal Center (Nangang, Taiwan). Mice were maintained in standard transparent polycarbonate cages (17.5 × 28 × 12.7 cm) with wire mesh tops in groups of five. BALB/cAnN-nude mice and severe combined immunodeficiency (SCID) mice (6–10 weeks, 20–30 g) were obtained from the National Laboratory Animal Breeding and Research Center (Nangang, Taiwan) and the National Taiwan University Laboratory Animal Center (Taipei, Taiwan), respectively, and maintained under specific pathogen-free conditions in groups of five in the Animal Care Center of Chang-Gung University (Taoyuan, Taiwan). The bedding was lined with shavings (Sani-Chips; Forest products, Montville, NJ, USA), which were renewed twice a week. Food pellets (Lab Diet 5010; LabDiet, Richmond, IN, USA) and water were provided ad libitum. The lighting was maintained on a 12 h light/dark cycle, the relative humidity varied between 50 and 60%, and the environmental temperature was maintained between 20 and 22oC. For tumour transplantation, anaesthesia was induced with an intraperitoneal injection of 1.625 mg/25 mg sodium pentobarbital (Somnotol; MTC Pharmaceuticals, Cambridge, Ontario, Canada). The animal experiment protocol was reviewed and approved by the Animal Ethics Committee of Chang-Gung University. At the end of the experiments, animals were euthanized with CO2 exposure.
Tumour cell lines
N-LMP1 (clone 1510) transfected BALB/c-3T3 (3T3/N-LMP1) cells (stable clones E2 and E9), and 3T3/Neo cells containing the vector alone were established as previously described by Chen et al. (1992). The E2-plus cell line was derived from ex vivo culture of an N-LMP1 E2 tumour. All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen Gibco BRL, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT, USA), 2 mmol/L
Induction and maintenance of the LMP1-dependent tumour model
To prepare tumour single cells (TSC), dissected tumour masses were minced into small pieces and then immersed in Hank's balanced salt solution (HBSS) medium (Invitrogen) containing collagenase D (1 mg/mL; Roche, Mannheim, Germany). Samples were incubated for 45–60 min at 37°C, ground and then filtered through a 53 µm nylon mesh (Spectra/Mesh, Houston, TX, USA) to exclude undigested tissue fragments. The resulting tumour suspension was centrifuged at 225 ×
Tumour histology
Tumour specimens were fixed in 10% formalin, embedded in paraffin and sectioned for light microscopy. The sections were stained with haematoxylin and eosin (H&E) using the standard procedures.
Western blotting analysis
Cells or tumour tissues were lysed at 95oC for 5 min in 100 µL of lysis buffer containing 60 mmol/L Tris–Hcl, pH 6.8, 2% sodium dodecyl sulphate (SDS; Sigma-Aldrich), 10% glycerol, 0.01% bromophenol blue (Merck, Darmstadt, Germany) and 0.1% mercaptoethanol (Sigma-Aldrich). The resulting protein extracts were separated by 10% SDS–polyacrylamide gel electrophoresis and electroblotted onto a nitrocellulose membrane (Amersham, Buckinghamshire, UK). For detection of LMP1 protein, the filters were incubated for 3 h with a mouse monoclonal antibody 40106, which is specific for the C-terminus of LMP1 (Fang et al. 2004). The filters were then washed three times with balanced salt solution containing 0.5% Tween 20 (Sigma-Aldrich), and incubated with an alkaline phosphatase-conjugated rabbit anti-mouse secondary antibody (Promega, Madison, WI, USA). The results were visualized by fast red salt precipitation in the presence of alpha-naphthyl phosphate (Sigma-Aldrich).
By periodic polymerase chain reaction (PCR) and Western blotting analysis, tumours stably expressed LMP1 protein along 31 tumour passages.
Polymerase chain reaction and DNA sequencing of LMP1
Genomic DNA was extracted from 1–2 tumour tissues (∼2000 mm3) which were collected from every 4–5 passages of the 31 in vivo tumour passages in three years. The DNA fragment(s) corresponding to the 3′ end of LMP-1 (at the position 168075–168390, including the 10-amino acid deletion region) was amplified using PCR with the sense primer (5′-AGCGACTCTGCTGGAAATGAT-3′) and the antisense primer (5′-TGATTAGCTAAGGCATTCCCA-3′) as previously described by Lin et al. (2001) in a GeneAmp PCR system 2400 or 9600 thermal cycler (Perkin-Elmer, Foster City, CA, USA). The reaction mixture consisted of 5 mmol/L Tris–HCl pH 8.0, 10 mmol/L NaCl, 0.01 mmol/L ethylene diamine tetra acetic acid, 0.1 mmol/L dithiothreitol (DTT), 5% glycerol, 0.1% Triton, 1.5 mmol/L MgCl2, 0.25 mmol/L dNTP, 10 pmol of primers, 1 unit of Taq DNA polymerase and 100 ng of genomic DNA from different origins. The temperature profile was 30 cycles of 30 s at 95°C, 30 s at 62°C and 1 min at 72°C, with a final extension for 10 min at 72°C. The amplified 286 bp fragment was separated on 1.5% agarose gel and recovered from gel using a GENECLEANII kit (BIO101) for nucleotide sequencing. The nucleotide sequences of all amplicons were determined at least twice in both sense and antisense directions.
Immunostaining for flow-cytometric analysis
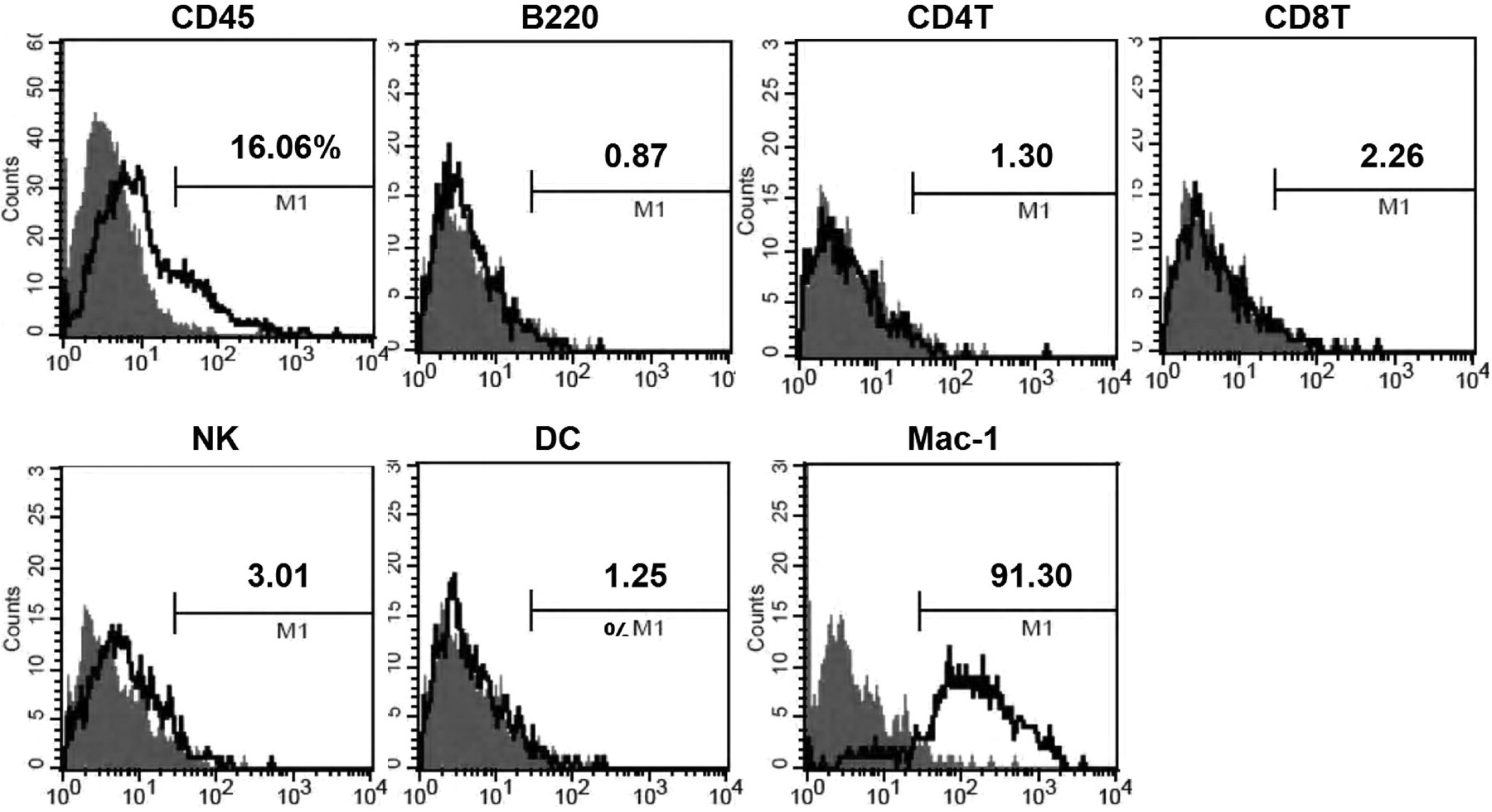
TSC or cell lines (106) were resuspended in 100 µL staining buffer (1% FBS and 1% NaN2 in phosphate-buffered saline [PBS]) with optimal dilutions of the appropriate antibodies and incubated for 30 min on ice. After incubation, cells were washed twice with cold staining buffer and analysed on a FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA). A total of 104 cells were analysed per sample, with dead cells excluded according to size and scatter. Characterization of leukocyte subsets in TSC was performed within CD45-positive gated cells. The utilized leukocyte subset markers were as follows: CD4, CD8 for T-cells, B220 for B-cells, DX5 for NK cells, CD11c for dendritic cells (DC) and CD11b for macrophages (Mac). Tumour cells were analysed within the CD45-negative population. The following conjugated monoclonal antibodies were obtained from BD PharMingen (San Jose, CA, USA): anti-CD45-CY (30-F11), anti-CD4-FITC (GK1.5), anti-CD8α-FITC (53-6.7), anti-B220-FITC (RA3-6B2), anti-DX5-FITC (DX5), anti-CD11c-FITC (HL-3), anti-CD11b(Mac-1)-FITC (M1/70) and anti-MHC I-FITC (36-7-5).
Results
Establishment of an N-LMP1 tumour model in BALB/c mice
Previous studies have shown that 3T3/N-LMP1 cells can form tumours in nude mice (Chen et al. 1992, Li et al. 1996, Yu et al. 2002). Here, we sequentially tested the use of three forms of 3T3/N-LMP1 cells (E2 and E2-plus cell lines and TSC) for establishing relatively efficient tumour growth in BALB/c mice. As illustrated in Table 1, E2 cells form tumours in all tested nude mice (n = 5), confirming the in vivo tumourigenicity of 3T3/N-LMP1. However, only two out of 41 (5%) of the normal BALB/c mice challenged with these cells developed tumours. Further use of TSC from the two tumour-bearing BALB/c mice only resulted in an 18% (9/50) tumour-take rate. To exclude the possibility that the low tumour incidence could be due to the induction of anti-tumour immunity by leukocyte infiltrates contained within the TSC, we tested both nude and BALB/c mice with E2-plus cells. All of the tested nude mice (5/5) developed tumours, but only five out of 14 (36%) BALB/c mice grew tumours. These results indicate that monodispersed tumour cells obtained from either culture or tumour masses were able to induce tumours in an immunocompetent environment, but with only a low tumour-take rate. Although the tumour incidence of the E2-plus cells was higher than that of E2 and TSC, this rate was still insufficient for establishing a stable tumour animal model (Table 1). In order to overcome the low tumour incidence in this system, we fragmented tumour masses and transplanted them into BALB/c mice. About 85% (223/264) of the mice developed tumours, suggesting that the presence of a preformed tumour mass is critical for N-LMP1 tumour induction in this immunocompetent host.
*Freshly prepared from the relatively rare tumour-positive BALB/c mice
In order to establish a reproducible protocol for tumour induction in BALB/c mice, we then tested whether tumour fragments obtained from immunodeficient nude or SCID mice could also serve as a stable tumour fragment source in BALB/c immunocompetent hosts. Our results revealed tumour-take rates of 85% (28/33) and 94% (43/46) in BALB/c mice receiving tumour fragments generated in nude mice using E2 and E9 cells, respectively (Table 2). In addition, E2-plus tumour fragments generated from either nude or SCID mice also showed tumour-take rates greater than 85% (Table 2). In this way, we were able to establish an N-LMP1 tumour mouse model in an immunocompetent host (Figure 1). The highest tumour-take rate was achieved by s.c. injection of N-LMP1-expressing cells into immunodeficient mice, followed by dissection of tumour fragments and transplantation of these tumour fragments into normal BALB/c mice. Afterwards, the N-LMP1 tumour mouse model could be maintained in vivo by grafting tumour fragments among normal BALB/c mice.
LMP1 = latent membrane protein 1
*Tumour fragments generated from nude or SCID mice were transplanted to BALB/c mice as described in Materials and methods
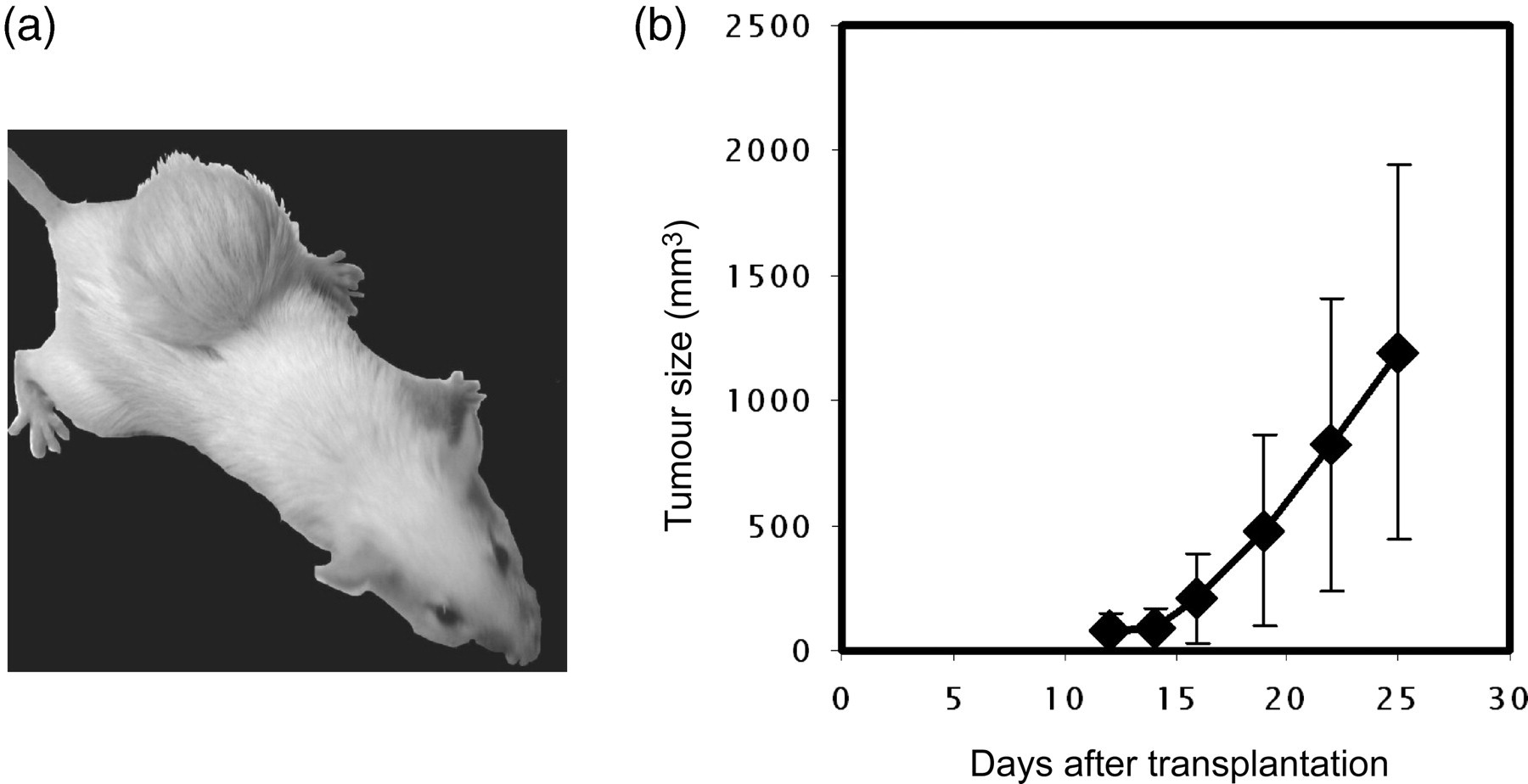
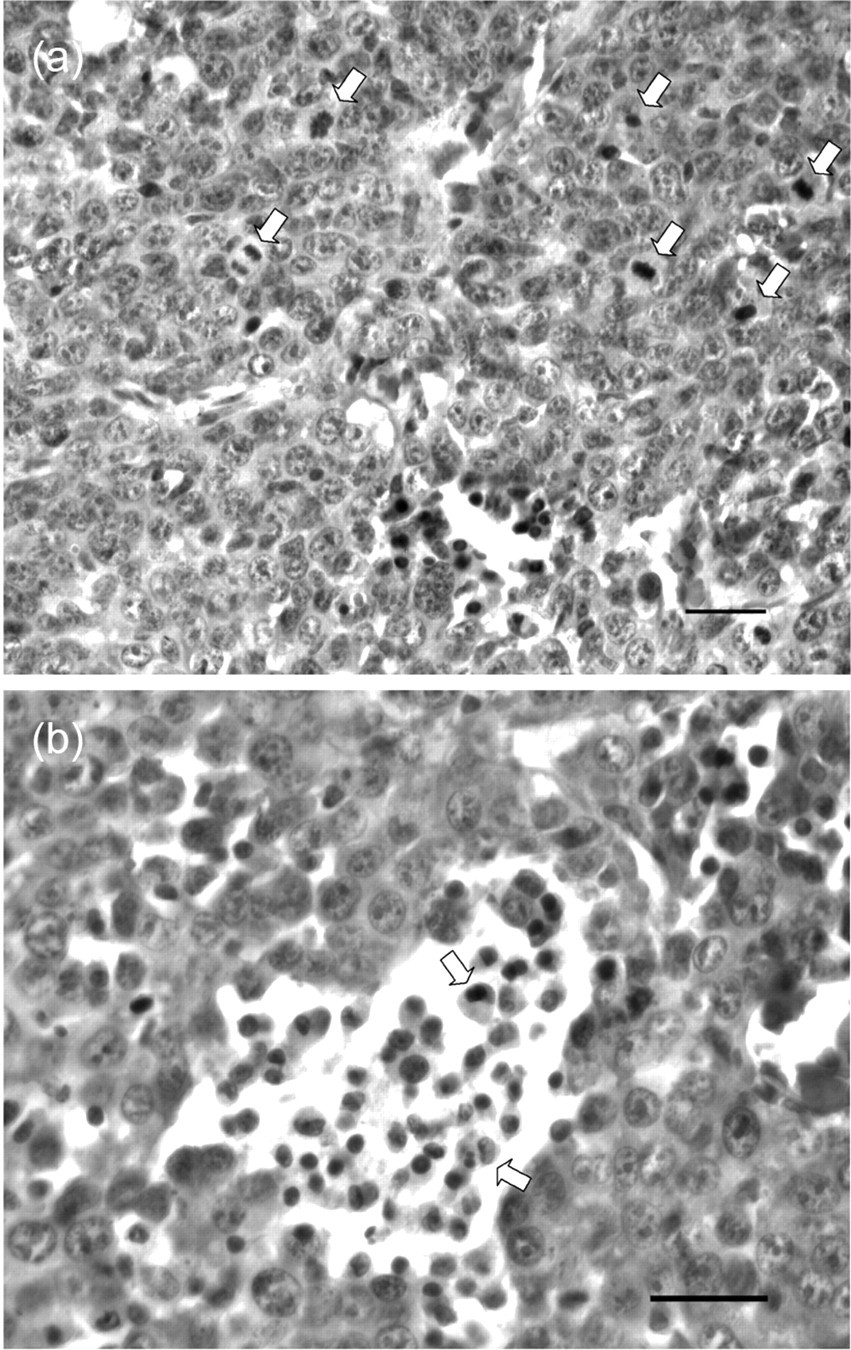
Representative tumours grown in BALB/c mice were photographed at week 3 (Figure 2a), and exponential growth curves were obtained by measuring the tumour volume and plotting as a function of time (Figure 2b). In general, tumours were palpable two weeks after transplantation, and progressive growth occurred in weeks 2–3. H&E staining was used to histologically examine tumours (Figure 3a). The results demonstrated that E2-derived transplanted tumour cells from BALB/c mice were spindle-shaped and arranged in sheets. Mitotic figures were readily identified (>20/10 high power field), confirming the malignancy of these N-LMP1-directed tumours. Furthermore, there were also leukocyte infiltrates present in the tumour periphery, where the highly proliferating tumour cells were located.
The presence of N-LMP1 and its stability in the tumour model
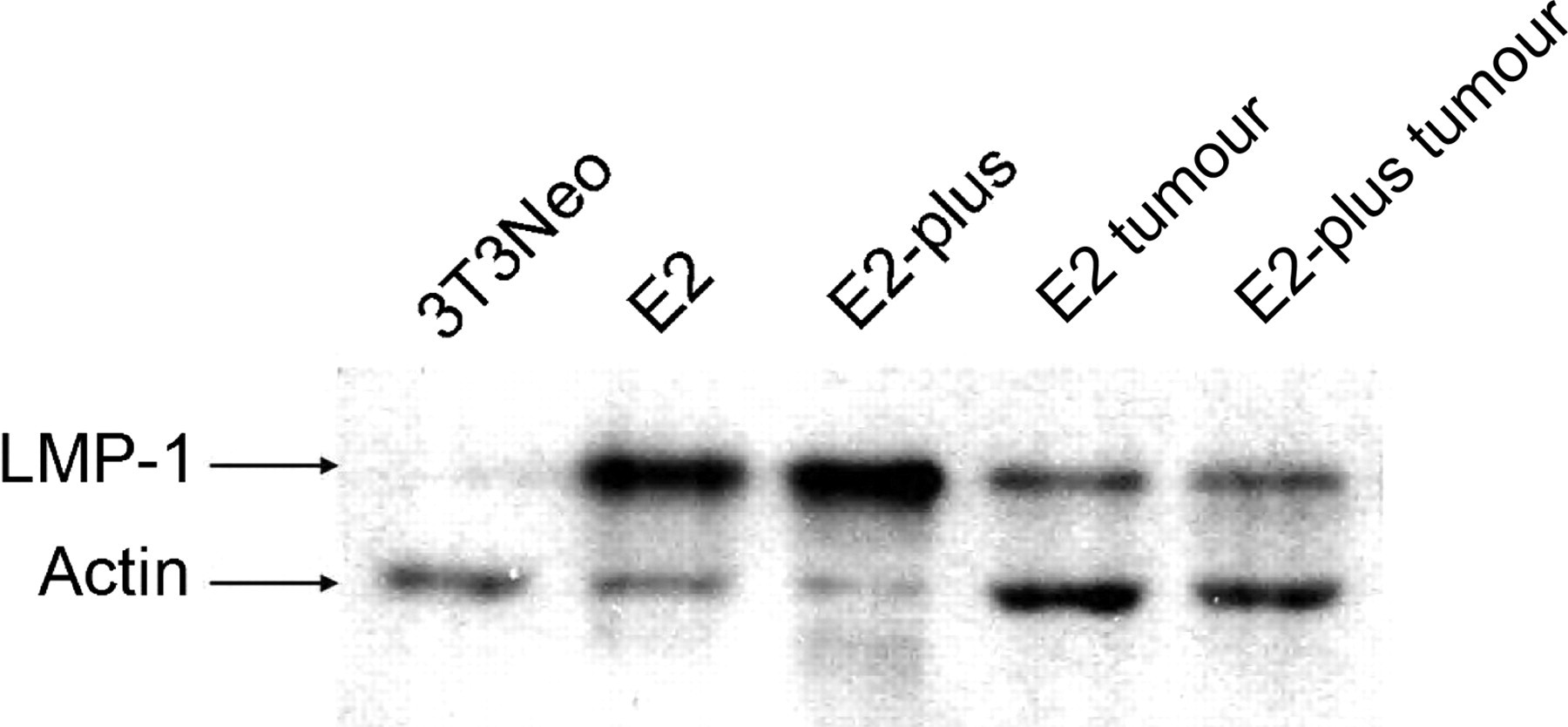
As persistent expression of N-LMP1 in the tumour mass is a critical feature for a usable animal model of this tumour type, we used Western blotting to examine the presence of N-LMP1 protein in transplanted tumour masses derived from E2 and E2-plus cells. As shown in Figure 4, no N-LMP1 proteins were detected in control 3T3/Neo. In contrast, detectable levels of N-LMP1 protein expression were found in E2, E2-plus cells and tumour masses derived from E2 or E2-plus cells in BALB/c mice, although the N-LMP1 protein levels in tumour masses were far lower than those in the corresponding cell lines. Furthermore, DNA variation has been frequently found in the LMP1 gene in NPCs of endemic regions (Miller et al. 1994), and the presence of amino acid substitutions and deletion of its C-terminal domain have been found to be important in cell growth, cell death and transforming ability (Li et al. 1996). Therefore, we further examined whether the immunocompetent environment might induce LMP1 variation within this region. DNA sequencing of tumour mass samples was performed during in vivo tumour passages, and the N-LMP1 gene sequences were compared with the previously published sequence (Chen et al. 1992). We found no detectable N-LMP1 gene mutations in our model system.
Infiltrates of N-LMP1 tumours in the model system
In order to examine why the tumour mass is critical for tumour establishment in the normal immunocompetent host, we used flow cytometry to analyse the types of leukocytes within the non-necrotic peripheral portion of the tumour mass. E2-derived tumour masses from BALB/c host were enzymatically digested into TSC, and the CD45-positive leukocyte populations were immunostained against markers specific for T-cells, B-cells, natural killer cells, DC and macrophages. Interestingly, the majority of the infiltrates were CD11b-positive (Figure 5), indicating that macrophages were the major cell type recruited by the N-LMP1 tumour. Moreover, the presence of macrophages in the tumour mass was also observed in the H&E-stained section (Figure 3b), suggesting the possible requirement of macrophages for N-LMP1 tumour establishment and maintenance.
MHC I expression by N-LMP1 tumours in the model system
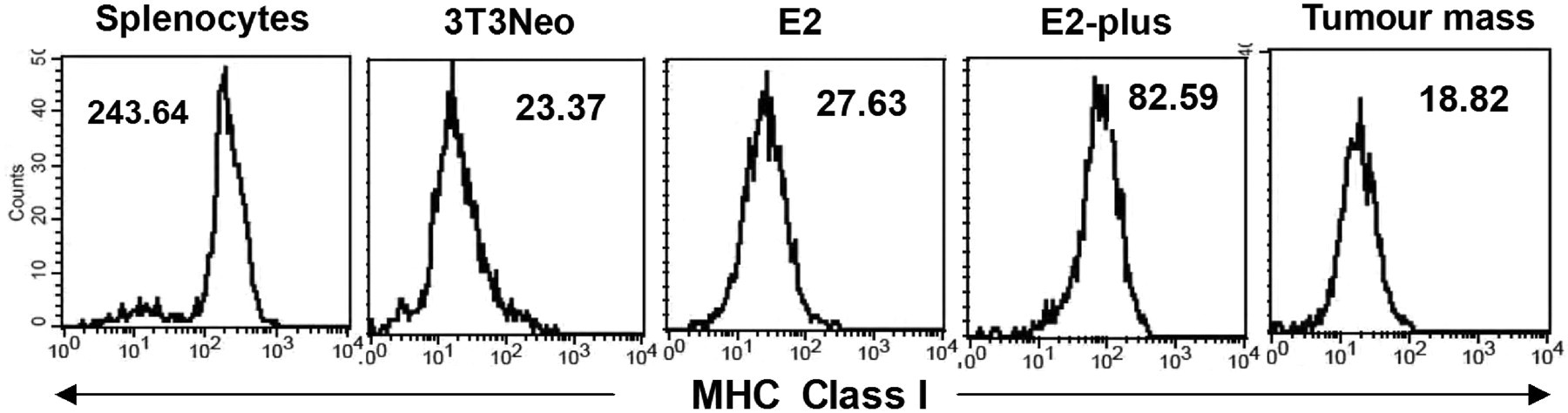
To examine whether the high rate of tumour development directed by tumour fragments in the immunocompetent host is due to decreased expression of MHC I on the tumour cells, we used flow cytometric analysis to determine the expression levels of MHC I on the 3T3/Neo, E2 and E2-plus cell lines and tumour cells (CD45-negative population) prepared from E2-derived tumour masses in BALB/c host. Although the CD45-negative population within the tumour may comprise non-leukocyte, non-tumour cells, such as stromal cells, endothelial cells, our results revealed that all aforementioned cells expressed significantly lower surface levels of MHC I than normal splenocytes (Figure 6). Furthermore, among those measured cells, E2-plus cells expressed approximately four-fold higher levels of surface MHC I than other cells. The latter finding may be partly due to the cell line, which was established from a long-term culture of TSC after removal of stromal leukocytes, indicating that the intact tumour mass may form an immunosuppressive microenvironment for the MHC I expression. From the above, we demonstrated that both N-LMP1 protein antigen and MHC I expression were down-regulated in the macrophages-infiltrating tumour mass, which may facilitate the tumour immune evasion leading to the high tumour-take rate via N-LMP1 tumour fragment transplantation.
Discussion
N-LMP1 has been shown to play a central role in the development of NPC in endemic regions. However, the study of N-LMP1-directed carcinogenesis has been somewhat limited by the lack of a simplified tumour animal model in an immunocompetent host. In this report, we present a novel N-LMP1 tumour model in immunocompetent mice (Figures 1, 2 and 3a). The tumours grown in BALB/c mice using this system are solely dependent upon N-LMP1 for oncogenesis, rather than involving the background complexity of a tumour cell line. Thus, our novel tumour model should facilitate the study of N-LMP1-directed tumour formation.
During the development of this model, we first attempted to directly inject E2 cells into immunocompetent BALB/c hosts. This effort was unsuccessful (Table 1). However, we were able to generate a low tumour incidence (5%) by injecting 3T3/N-LMP1 cells into the BALB/c mice. It is formally possible that this low tumour-take rate could be due to poor survival during transfer between the in vitro and in vivo systems, but our finding that these cells were highly tumourigenic when injected into nude mice seemed to rule out this possibility. The considerably lower tumour incidence in the immunocompetent vs. immunodeficient hosts may suggest the participation of T-cells in N-LMP1 tumour prevention. This possibility is further supported by the slightly increased tumour-take rate of in vivo-adapted E2-plus cells (36%) in BALB/c mice. Previous studies have shown that s.c.-inoculated monodispersed cancer cells can directly migrate into the draining lymph nodes to induce CTL responses, resulting in tumour rejection (Ochsenbein et al. 1999, 2001, Spiotto et al. 2004). In addition, the two-fold lower tumour-take rate of TSC (18%) vs. E2-plus cells may further reflect the presence of immune-triggering leukocytes in the TSC, consistent with previous reports demonstrating that the migration of the stromal leukocytes carrying tumour antigens into the draining lymph nodes leads to activation of the CTL response (Singh et al. 1992, Spiotto et al. 2002, Yu et al. 2006). These results revealed that the s.c. injection of monodispersed N-LMP1 tumour cells may be immune-stimulatory, and could not serve as an appropriate way to establish stable N-LMP1 tumour model in the immunocompetent host.
We achieved a much higher tumour-take rate with N-LMP1 tumour fragments implanted in BALB/c hosts (>85%) demonstrating that immune acceptance was greatly improved through the use of a preformed tumour mass (Table 2). The mechanisms responsible for tumour fragment survival in an immunocompetent host have not yet been fully elucidated. One possibility is that stromal confinement prevents the cancer cells and/or the leukocyte infiltrates from migrating, leading to decreased induction of the CTL response in draining lymph nodes (Speiser et al. 1997, Ochsenbein et al. 1999, 2001). Another explanation is that the presence of stroma produces a microenvironment that favours tumour expansion. This possibility is supported by a recent report that tumour-associated macrophages are key regulators in the process of stromal formation (Mantovani et al. 2002, Pollard 2004), and by our observation that the primary infiltrating leukocyte population of the transplanted tumours consisted of macrophages (Figures 3b and 5). The third possibility is that the organization of the stroma may provide a strictly regulated microenvironment that allows tumour cells to avoid immune attacks delivered by infiltrating CTLs. Our observation that the N-LMP1 and surface MHC I expression levels were far lower on TSC tumour cells vs. E2-plus cells (Figures 4 and 6) suggests that local immunosuppression may contribute to tumour survival in our model system. This notion is also consistent with previous findings that stromal formation induced systemic immunosuppression via tumour-associated macrophages (Mantovani et al. 2002). Collectively, our findings and the previous reports suggest that the tumour-take rates of monodispersed cancer cells or tumour fragments in immunocompetent mice is primarily determined by whether or not an effective immune response can be initiated in the draining lymph node, and further suggest that successful formation of tumour stroma may provide tumour growth advantages, even in an immunocompetent environment.
Thus, we herein describe the development of an N-LMP1-derived tumour model in an immunocompetent host. We then further showed that our model maintains stable expression of N-LMP1 in the tumour mass throughout in vivo tumour passages (Figure 4). This suggests that N-LMP1 expression may be essential for both tumour initiation and maintenance. Furthermore, no mutation(s) in the C-terminal effector domain of the expressed N-LMP1 was found in our system, suggesting that immune pressure may not lead to LMP1 gene variation and/or that the continued presence of stroma in the in vivo tumour passages in our model may protect the tumour cells from immune attack, alleviating the need for mutation(s). This consistent expression of un-mutated N-LMP1 in tumour mass-derived tumours in an immunocompetent host indicates that the new model system herein may be useful for future studies of N-LMP1 immunogenicity and possible therapeutic modulation of NPC in endemic areas.
In conclusion, we herein report the establishment of a simplified oncogene-directed tumour model using BALB/c-3T3 transfectants in an immunocompetent mouse host. The oncogene-dependent transformation of the system means that the cellular background of the grown tumour is much simpler than that of the models established by gene transfection into pre-existing tumour cell lines. This novel approach provides a direct in vivo assessment system for oncogene research, and may serve as a platform for evaluating future oncogene-based tumour therapies.
Footnotes
Acknowledgements
This work was supported in part by Medical Research Grant CMRP725 from the Chang-Gung Memorial Hospital, Striving for Excellence Grant 89-B-FA04-1-4 from the Ministry of Education, Taiwan, a grant (NSC85-2331-B-182-112-M22) from the National Science Council of ROC and a grant to the Functional and Micro-Magnetic Resonance Imaging Center (NSC93-3112-B-001-006-Y) from the National Research Program for Genomic Medicine, National Science Council of ROC. The authors thank C N Ou-Young for excellent technical assistance, and Drs S K Liao for helpful discussions, C J Huang for assisting with tumour transplantation techniques, H C Hsu, Y M Jeng and W C Liou for advice and assistance with histology, and Y C Chien and P Tang for assisting with the DNA sequencing.