
Abstract
Objective
To assess the frequency of the D1152H mutation in the CFTR gene in normal individuals, in cystic fibrosis (CF) patients and in the setting of prenatal diagnosis.
Setting
A database analysis of sequential screening results seen at the Sheba Medical Center, Israel, between 2001 and 2010.
Methods
We retrospectively analyzed the frequency of D1152H in a large cohort of healthy individuals who were screened as part of a routine prenatal care programme, in individuals referred due to CF-related symptoms and in the setting of prenatal diagnosis.
Results
We found one asymptomatic homozygous female and 195 D1152H carriers among 49,940 healthy individuals screened, establishing a carrier rate of 1:255 for this mutation. We detected D1152H in nine of 103 individuals referred due to CF-related symptoms. Four suffered from respiratory symptoms and five from congenital bilateral absence of the vas deferens (CBAVD). During this period D1152H was detected in three pregnancies, two of which were aborted.
Conclusion
The increased frequency of D1152H in individuals referred due to CF-related symptoms compared with healthy individuals included in the CF carrier screening programme (P < 0.001) clearly indicates that it is a disease-causing mutation.
Introduction
The D1152H mutation is associated with residual CFTR function and abnormal chloride gating. 10 Initially this mutation in its homozygous form or in conjunction with another CFTR mutation was associated mainly with CBAVD and mild, late onset pulmonary disease; with borderline or normal sweat chloride values.7,11 Moreover, among Hispanics D1152H was found in 6% of the referrals for routine screening but in no Hispanic CF patients, questioning whether D1152H is no more then a functional polymorphism; 12 and in a recent consensus report D1152H was not categorized as a disease-causing mutation. 13
We have included D1152H in our CF carrier screening programme and in patient screening since 2001. During these years we have detected D1152H in healthy individuals who took part in our screening programme, in patients, and in three fetuses studied prenatally. In this report we summarize our experience with this mutation.
Methods
The study was performed at the Sheba Medical Center, Israel, after being approved by the institutional review board. Between 2001 and 2010, the CF population screening evolved as described in Table 1. Molecular diagnostics were performed with three different kits: ElucigeneTM CF7 and CF-MEP (OrchidBioScience, UK) during 2001–2008, Tag-ItTM mutation test detections kit CFTR version 2.01 (Tm Bioscience, Toronto) during 2006–2007 and Nanogen CF-14, Gamidor Diagnostics Ltd, Israel since 2008. In addition, four mutations were diagnosed by RFLP and two deletion mutations by ABI genetic analyzer (details will be supplied upon request). Clinical data were obtained from the medical files.
List of mutations performed according to dates
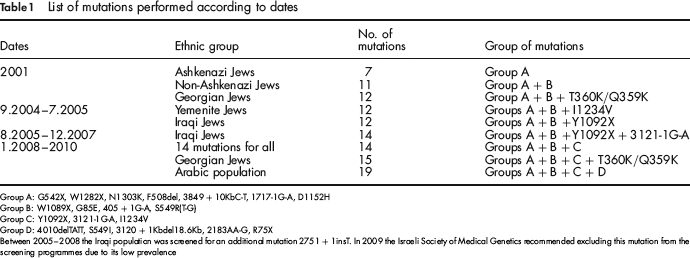
Group A: G542X, W1282X, N1303K, F508del, 3849 + 10KbC-T, 1717-1G-A, D1152H
Group B: W1089X, G85E, 405 + 1G-A, S549R(T-G)
Group C: Y1092X, 3121-1G-A, I1234V
Group D: 4010delTATT, S549I, 3120 + 1Kbdel18.6Kb, 2183AA-G, R75X
Between 2005-2008 the Iraqi population was screened for an additional mutation 2751 + 1insT. In 2009 the Israeli Society of Medical Genetics recommended excluding this mutation from the screening programmes due to its low prevalence
Results
Between January 2001 and June 2010 we screened 49,940 individuals for CFTR mutations and found 1524 carriers, implying a carrier rate of 1:33 (Table 2). One hundred and ninety-five individuals were found heterozygous for the D1152H mutation and one 30-year-old female without any CF-related symptoms was found homozygous for D1152H. The carrier rate of this mutation was 1:255, comprising 12.8% of the mutations detected.
Carrier screening for CF between 2001–2010

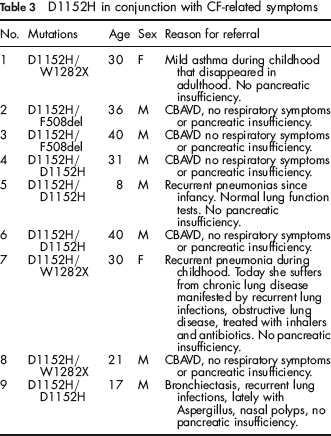
During this time period 103 individuals were tested because of CF-related clinical symptoms. Thirty-seven patients were found to have two mutations, nine were found to carry one mutation and in the rest no mutations were found. The relatively low mutation detection rate among this cohort is most probably due to the fact that not all of them had CF and reflects a low threshold for sending suspected patients for mutation analysis among the referring physicians. Of the 37 patients with two mutations, D1152H was found in nine (Table 3), comprising 16.9% of the mutations detected among suspected patients. Four were homozygous for D1152H, and five were compound heterozygotes. Two of the D1152H homozygotes were referred because of CBAVD and two due to respiratory symptoms. Three of the compound heterozygotes were referred because of CBAVD and two due to respiratory symptoms. D1152H was not detected in any of the nine patients with a single mutation.
D1152H in conjunction with CF-related symptoms
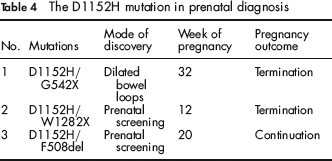
Between 2001 and 2010 we detected three D1152H compound heterozygote fetuses (Table 4). The parents of the first fetus did not perform the CF screening test and were referred due to dilated bowel loops on US. In the other two cases, prenatal diagnosis followed the screening tests of the parents who were found to be carriers. The mother of the third fetus was also a carrier of a risk allele for Huntington chorea (HC). The fact that her fetus inherited the normal HC allele probably influenced her decision to continue the pregnancy.
The D1152H mutation in prenatal diagnosis
Discussion
In this report we present our experience with the D1152H mutation in a low-risk healthy population screened for CF carrier state, in symptomatic referrals and in prenatal diagnosis. Over representation of a mutation in patients, compared with the general population is considered one of the cornerstones in differentiating a mutation from a benign polymorphism. We found a much higher frequency of the D1152H allele in individuals referred due to possible CF (13 of 206 alleles) compared with individuals who took part in the screening programme (195 of 99,880; P < 0.0001). These results stand in sharp contrast to those of Sugerman et al., who found D1152H in 6% of the Hispanic population but not in Hispanic CF patients. 12 A carrier rate of 1:255 for D1152H combined with a general carrier rate of 1:33 implies that roughly 1:33,000 individuals is a compound heterozygote or homozygote for D1152H, a figure in line with the single asymptomatic homozygote that we have detected in our screening programme. In this study, the overwhelming representation of D1152H compound heterozygotes and homozygotes in the patient cohort (9 of 103) serves as additional evidence that D1152H is associated with clinical symptoms thus supporting two recent publications. In 2006 Mussaffi et al. described nine patients, two homozygous for D1152H and seven compound heterozygotes, aged eight months to 54 years with variable CF manifestations which included lung and pancreatic involvement, and CBAVD. 14 Although the symptoms were often mild, the authors concluded that D1152H-associated lung disease can appear in early infancy and in adults diagnosed at a late age can be quite severe. Burgel et al. described 42 patients with the D1152H mutation; 80% presented with respiratory symptoms and 25% with CBAVD. 15 Clinical manifestations among patients presented in our study are similar to those reported in the two previous series. Four of our patients presented with respiratory symptoms and although one was diagnosed at the age of eight months, all of the others were diagnosed at an older age, indicating that D1152H is associated with a relatively mild disease. Five male patients in our cohort presented with reproductive problems. Mussaffi et al. found that two of the three adult males in their study had children, suggesting that homozygosity or compound heterozygosity for D1152H in males does not invariably result in infertility. It is important to emphasize that male CF-related infertility can be successfully treated by in vitro fertilization.
The atypical disease and relatively mild symptoms pose an ever increasing counselling problem when faced with a fetus homozygous or compound heterozygous for D1152H as exemplified by the three cases described above. Counselling was given by a genetic counsellor and an experienced physician from the CF clinic at Sheba Medical Center, who described to the couples the possible clinical spectrum. Two of the couples decided to terminate the pregnancies while one couple decided to continue. In the first case, the finding of dilated bowel loops on a routine second trimester ultrasound examination prompted a CF carrier workup in the parents and consequently a genetic diagnosis in the fetus. Dilated bowel loops in a fetus with CF results from bowel obstruction by a meconium plug and is usually associated with a bad prognosis. In contrast to the decision made by these parents, Mussaffi et al. described a fetus with dilated bowel loops and the D1152H/F508del genotype, where the parents elected to continue the pregnancy. 14 Normal meconium was passed 16 hours after birth and subsequent studies did not reveal pancreatic insufficiency, although long-term follow-up revealed mild pulmonary symptoms and hyperinflation on a chest X-ray. In the third case, there is no doubt that the normal testing for Huntington in the fetus in view of the carrier state of the mother played a major role in the couple's decision to carry on with the pregnancy.
Aside from the severity of the disease, the decision to continue or terminate the pregnancy in such circumstances relies on numerous factors and includes the social, religious and ethnic background of the couple, the number of children at home and difficulties encountered in getting pregnant. Although the final choice is made by the parents, the personal views and beliefs of the genetic counsellors and the medical team, which are passively conveyed to the parents throughout the counselling process, influence their final decision. Hopefully, more information on the nature and prognosis of patients harbouring this mutation will help couples decide the fate of the pregnancy when a fetus homozygous or compound heterozygous for this mutation is detected.