
Abstract
Objective
To expose causes leading to the delayed arrival of phenylketonuria (PKU) patients at a governmental reference centre (RC), and to describe their clinical characteristics.
Material and methods
PKU files registered during the past 18 years at the National Institute of Pediatrics in Mexico City were evaluated. Patients were classified into two groups according to their age at arrival: Group I (early reference), patients arriving during the first month of life; and Group II (late reference), those who arrived after thirty days of age. Time and causes of delay were documented.
Results
Of 57 recorded files, 10 were classified in Group I and 47 in Group II. Causes leading to the late arrival of Group II patients were absence of routine newborn screening (NBS), PKU not included in the routine NBS, sampling after the recommended age, false negative result, results without interpretation and/or instructions to follow, delayed notification of results, poor medical criteria of attending physician, difficulties in obtaining confirmatory tests, and administrative failures.
Conclusion
The main cause of late referral of PKU patients was the absence of PKU testing. As a developing country, Mexico still faces challenges in the proper functioning and expansion of the NBS programme. Most PKU patients arrived at the RC late, presenting with varying degrees of the clinical spectrum. Incorporating PKU testing into the already established Mexican NBS system and adding quality indicators to guarantee proper operation in all NBS phases is necessary to achieve the goal of identifying, referring, diagnosing, and treating patients promptly.
Introduction
Without treatment, most patients will develop mental retardation. It is well known that adequate treatment of infants with PKU within the first two weeks of life substantially improves their outcome and promotes normal growth and development.2–5 Because of this, early diagnosis in the first days of life is essential. Mass screening of PKU by newborn screening (NBS) is a public health practice that has been carried out for the last 50 years. 6 Nowadays, many countries around the world have national NBS programmes for PKU. Based on substantial evidence, the US Preventive Services Task Force (USPSTF) reaffirmed screening for PKU as a grade ‘A’ recommendation, which means there is high certainty that the net benefit is substantial. 7 The cost-benefit ratio for PKU screening has been analyzed in Mexico and other countries.8–13
More than 15 years ago, Velazquez et al. demonstrated the feasibility and necessity of PKU NBS in Mexico 14 , but despite his efforts, PKU NBS is not mandatory. Therefore, this disease is still a cause of mental retardation in the Mexican population.
The mandatory NBS programme in Mexico only includes congenital hypothyroidism (CH). Although the coverage of public NBS programmes for CH has made significant progress 15 , the inclusion of other genetic disorders like PKU has been left behind. 16 Currently, there is huge variability between NBS practices in Mexico; only a few public health institutions include PKU NBS, whereas children treated at other health-care facilities do not have access to this testing. 17
On the other hand, it is well known that patients with complex rare diseases like PKU have better outcomes when they are treated in third level health-care reference centres (RC). 18 The European Rare Diseases Organization defines a RC as the place that should provide multi-disciplinary patient-centred care, and must be managed in a co-ordinated way. It should offer a reliable and accurate diagnosis, as well as genetic testing and counselling, and access must be provided to all patients regardless of their country or region of origin. 19
According to the established guidelines of the Mexican Ministry of Health, patients must be sent from one healthcare level to another through a well known system of reference and counter reference, and patients with inborn errors of metabolism like PKU must be treated at third level healthcare centres.
Despite the fact that our facility meets the above criteria, we observed delays in the arrival of patients and the clinical consequences. The aim of this study was to expose the main causes leading to the delayed arrival of PKU patients at a RC, and to describe their clinical and biochemical characteristics at the time of their arrival.
Methods
This study was approved by the institutional review board of the National Institute of Pediatrics (INP), number 30/2010. This retrospective and descriptive study was carried out at the Inborn Errors of Metabolism and Screening Laboratory (LEIMyT) of INP in Mexico City, which is a governmental RC for inherited metabolic disorders and receives patients from medical institutions all over the country.
All PKU files registered during the period from January 1992 to December 2010 were evaluated. The patients were classified into two groups according to their age at the time of arrival at the RC. These were Group I: early reference; patients who came during the first month of life, and group II: late reference; those who arrived at the RC after thirty days of age. Because of the lengthy study period, patients were stratified according to their date of birth, which was related to the status of NBS in Mexico at different time periods.
The time of delay was recorded and defined as the age at arrival minus the optimal time of referral (30 days old). In Group II, the causes of delay were documented following the known NBS phases; preanalytic (education and screening), analytic (laboratory testing), and postanalytic (reporting, short-term follow-up/tracking, diagnosis, treatment/management, ancillary services, and outcome evaluation) activities. 20 A positive diagnostic confirmatory test was based on Phe and Tyr concentrations as quantified by high performance liquid chromatography (HPLC) 21 , with Phe >240 μM and a Phe/Tyr ratio >2.5 considered positive. 22
Regardless their age, all patients started treatment once the diagnosis was confirmed at the RC according to the institutional care model, and all the affected families received professional genetic counselling. 23
Geographic origin, gender, consanguinity, number of affected siblings, and clinical data at the moment of arrival were analyzed; the latter were classified by organ system as digestive, dermatologic and/or neurologic. Mental retardation scores were determined according to the clinical description of the tenth version of the World Health Organisation International Classification of Diseases (ICD-10) 24 based on the available information in each file.
Results
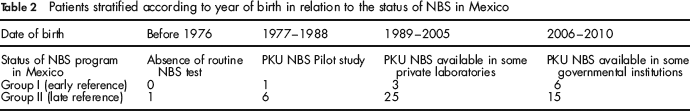
We studied 57 files. Ten were classified as Group I (early reference) and 47 as Group II (late reference). Gender, age and biochemical characteristics are shown in Table 1. Subjects came from 50 families. Seven families had two affected siblings, whereas consanguinity was documented in four families, including one of the families with two affected siblings. The geographic origin of the families is shown in Figure 1. Table 2 displays the number of patients who arrived at the RC, stratified according to their date of birth in relation to the NBS status in Mexico at that time. All patients in Group I were sent to the RC because of a suspicious NBS result, and all patients were asymptomatic.
General characteristics of the studied patients stratified by age upon arrival at the RC

Patients stratified according to year of birth in relation to the status of NBS in Mexico
Geographic origin of the PKU families. *One family with two affected siblings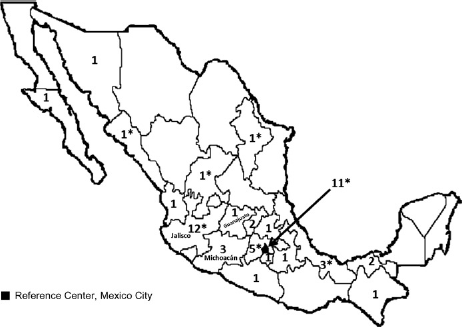
Of the 47 subjects in Group II, 29 were referred by physicians who suspected an inborn error of metabolism based on clinical data, 17 arrived because of a suspicious NBS result, and one was referred by the patient's father, who recognized the clinical picture based on his own experience. The delays in arrival ranged from two days to 16 years and five months. Eleven patients in Group II were found to be asymptomatic, and the remaining (36) showed clinical signs of the disease (Figure 2).
Clinical manifestations observed in symptomatic patients (n = 36) from group II at the time of arrival at the RC. *Includes hyperactivity, stereotypy, aggressiveness, anxiety and/or social withdrawal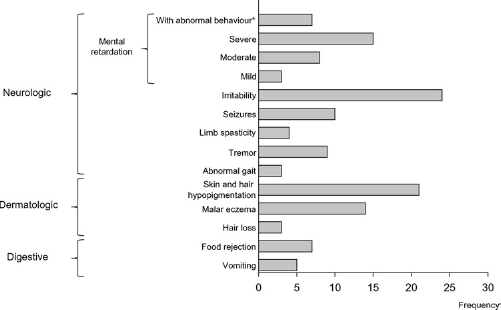
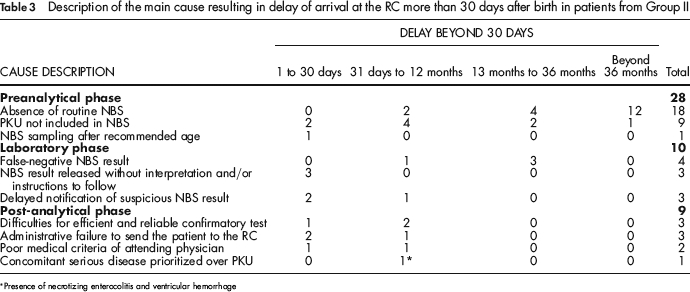
The causes leading to the late arrival of the patients in Group II were as follows: absence of routine NBS, PKU not included in the NBS, sampling after the recommended age, a false-negative result (true PKU patient with a normal NBS result), verbal notification or printed results without interpretation and/or instructions to follow, delayed notification of suspicious results (after 30 days of age), poor medical criteria of the attending physician (includes forthright abnormal results sent for analytical NBS reprocess two or more times instead of being promptly notified of the confirmatory test), difficulties in obtaining efficient and reliable confirmatory tests (blood Phe quantification by HPLC), and administrative failure to send the patient to the RC. The main causes of delay in each patient in Group II are summarized in Table 3.
Description of the main cause resulting in delay of arrival at the RC more than 30 days after birth in patients from Group II
Presence of necrotizing enterocolitis and ventricular hemorrhage
Discussion
This study documents the causes leading to the late arrival of PKU patients to the RC. Notably, 82% of the patients studied arrived late, with a mean age of two years and eight months; however, the oldest patient was 16 years old at the time of his arrival (Table 1). The main cause of delay was the absence of screening for PKU in 27/47 cases; of these patients, 18 had no routine NBS and PKU testing was not included for nine patients even when the NBS was performed. These striking data show that despite advances in the NBS programme for CH in Mexico, 18/57 (32%) of patients in this cohort did not have access to routine NBS.
CH and PKU tests can be performed from the same blood sample and at the same facility. Regardless of the available methodology (fluorometric analysis or tandem mass spectrometry), this early opportunity is possible because a number of biomarkers for several metabolic disorders can be detected from a single dried blood spot specimen.22,25 Not to do so is a misuse of the complex NBS logistic that requires sampling, handling and shipping those samples, as well as a number of human resources that are involved in the process.20,26
In five cases, the cause of delay was inappropriate time of NBS sampling, which is a preanalytical problem. The optimal time for collecting the sample for NBS is between the first three and five days of life. 27
Regarding the analytical phase, we found four false negative cases, which constitute a severe failure in the NBS. All these cases were symptomatic patients who arrived after more than three years of delay; this suggests that inadequate analytical processes misled the differential diagnosis of PKU. In the retrospective analysis of these cases, we found that all of these patients were screened by thin layer chromatography, a method that is no longer used in modern screening laboratories because of its poor sensitivity, i.e. Phe blood levels below 480 μM cannot be consistently detected with this method. 28 In this population, the most frequent errors occurred in the post-analytical NBS phase. It is important to note that this phase is a complex process involving the actions of multiple individuals with different criteria, including biochemists, nurses, physicians and social workers, among others. The greatest number of mistakes was documented in patients who had NBS and arrived between one and 30 days late (Table 3). Although this delay was apparently not excessive, it is known that time and exposure to high blood concentrations of Phe negatively impacts children's neurodevelopment. 29
In addition to the main reasons for delay for each patient listed in Table 3, it is important to mention that some patients experienced more than one cause for their late arrival, and four mistakes were recorded for one patient. While a single error may not cause significant delay in the arrival of patients, it is the sum of the events that cause disruption to the NBS process.
In an 11-month-old boy, a serious concomitant disease was prioritized; this child died of respiratory failure as a complication of pneumonia. He was a twin who suffered from twin-twin transfusion syndrome, necrotizing enterocolitis, and intracranial hemorrhage with hydrocephalus. His initial NBS specimen showed a Phe concentration of 660 μM. Because of his condition, the confirmatory diagnosis was established when the boy was six months of age, with Phe 1508 μM and a Phe/Tyr ratio of 12.84. Even when the patient dies, genetic counselling should be offered so the parents can make informed reproductive decisions.
The mean time of reference in Group I was 18 days (Table 1); this period could be optimized in order to begin nutritional management within the first two weeks of life.2,4,28 Concerning the geographical origin of the patients, we cannot make epidemiological assumptions, but nearly 35% of the patients came from the western area of the country, which includes the states of Jalisco, Guanajuato, and Michoacán (Figure 1). The hypothesis that there is a higher PKU prevalence in this area was published several years ago, but has not been confirmed. 30
The clinical data described in Figure 2 are similar to the natural history of the disease reported in the literature, mostly affecting the central nervous system. 1 Contrary to what happens in developed countries, where PKU has grown from a symptomatic disease to a genetic condition modified by early treatment,31–33 we still treat patients with the whole phenotypic spectrum. Meanwhile, all physicians who treat patients with mental retardation and or behavioral problems should rule out PKU in the differential diagnosis.
All processes are subject to error, especially those involving multiple personnel such as NBS. In order to improve this programme in countries where NBS is still under development, the following strategies are suggested: inform parents and primary care physicians about the benefits of NBS; provide health personnel with ongoing training in all phases of NBS to achieve proper and universal sampling between the first three to five days of life; and supply clear instructions for all abnormal results along with fact sheets and printed results with directions to follow. All parents should be aware of the existence of PKU and must have access to a specialized RC so as to obtain efficient and reliable confirmatory tests. Because of this, most countries have quality control systems for NBS, which helps to guarantee its performance.20,34 Our findings highlight the need to implement comprehensive quality evaluation in the NBS programmes in Mexico.
Conclusion
The main cause of late referral of patients with PKU was the absence of a PKU test. PKU is the paradigm of inborn errors of metabolism that led to the creation of a mass screening test (NBS) almost 50 years ago, with the main objective of avoiding the neurological complications of the disease. However, as a developing country, Mexico still faces problems and challenges in the proper functioning of the NBS, as well as its expansion to diseases other than CH. As a result, most PKU patients came late to our RC, presenting with varying degrees of the clinical spectrum. It is necessary to incorporate PKU testing in the already established NBS system, and to add quality indicators to guarantee proper operation in all phases of the system to achieve the goal of identifying, referring, diagnosing, and promptly treating patients.