
Abstract
Objectives
It has been suggested that homozygous c.985A>G medium-chain acyl-CoA dehydrogenase deficiency (MCADD) is a disease of White ethnic origin but little is known regarding its ethnic distribution. We estimated ethnic-specific homozygous c.985A>G MCADD birth prevalence from a large-scale UK newborn screening study.
Methods
Homozygous c.985A>G MCADD cases were ascertained in six English newborn screening centres between 1 March 2004 and 28 February 2007 by screening approximately 1.1 million newborns using tandem mass spectrometry analysis of underivatised blood spot samples to quantitate octanoylcarnitine (C8). Follow-up biochemistry and mutation analyses for cases (mean triplicate C8 value ≥0.5 µmol/L) were reviewed to confirm diagnosis. Ethnicity was ascertained from clinician report and denominators from 2001 UK Census estimates of ethnic group of children less than one year.
Results
Sixty-four infants were c.985A>G MCADD homozygotes (overall prevalence 5.8 per 100,000 live births; 95% CI 4.4–7.2). Sixty (93%) were White, two (3%) were mixed/other and two were of unknown ethnic origin. No Asian or Black homozygotes were identified. Proportions of White, mixed/other, Asian and Black births in screening regions were estimated, yielding homozygous c.985A>G MCADD birth prevalence of 6.9 per 100,000 (95% CI 5.2–8.8) in White, and 95% CI estimates of 0–2.7 per 100,000 in Asian and 0–5.8 in Black populations. The c.985A>G carrier frequency in the White group was estimated at one in 65 (95% CI 1/74, 1/61) under Hardy–Weinberg conditions.
Conclusion
c.985A>G homozygous MCADD is not found in Black and Asian ethnic groups that have been screened at birth in England. This is consistent with the earlier published observations suggesting that MCADD due to the c.985A>G mutation is a disease of White ethnic origin.
INTRODUCTION
Individuals affected by medium-chain acyl-CoA dehydrogenase deficiency (MCADD) may present with hypoglycaemia progressing to lethargy, encephalopathy or sudden unexpected death, often precipitated by fasting or illness. 1 MCADD is an autosomal recessive metabolic defect of fatty acid oxidation due to the reduced activity of the medium-chain acyl-CoA dehydrogenase (MCAD) enzyme (EC 1.3.99.3). Among those who experience acute episodes, the risk of death is estimated to be 25%. 2–4 As some children remain asymptomatic, the overall cohort mortality is lower at 12–20%. 5,6
Screening aims to identify infants with MCADD and prevent the onset of symptoms by early dietary management to reduce adverse outcomes of intellectual disability and death. The overall birth prevalence is one per 10,000 live births; approximately 80% of clinically diagnosed MCADD cases are homozygous for the common c.985A>G mutation, however this percentage is lower (approximately 45–55%) in cases ascertained through newborn screening. 5 It has been shown that the frequency of homozygosity for the common mutation is substantially higher in clinically presenting cases than in those who are screened as screening identifies MCADD deficiency with twice the frequency than in an unscreened cohort, among those with mutations which may, or may not, present clinically. 5
It has been suggested that this common mutation is only found in patients of European ancestry. In 1993, Gregersen et al. 7 reported a Northwestern European founder effect for the c.985A>G mutation, thus proposing that MCADD, due to this mutation, is exclusively a disease affecting those of white ethnic origin. Small-scale studies in non-White populations in the US indicate that the prevalence of the c.985A>G mutation may be much lower among persons of non-White ancestry relative to persons of European descent; however, robust estimates of c.985A>G prevalence have not been reported to date, possibly reflecting the absence of screening in populations of predominantly non-White ethnicity as well as failure to report prevalence by ethnicity in the existing large-scale studies. 8,9 Previous work by Seddon et al. 10 reported a UK-wide c.985A>G carrier frequency of between 1/52 and 1/83 but did not report these data by ethnic group. Despite a recent increase in the publication of a number of MCADD screening studies, there has been no report of the ethnic distribution of homozygous c.985A>G MCADD, which might substantiate earlier findings of a Northwestern European founder effect.
We estimated the prevalence of homozygous c.985A>G MCADD and c.985A>G carrier frequency by ethnic group, as part of a large-scale multicentre prospective UK study of universal newborn screening for MCADD. Importantly, the population screened in this study is ethnically diverse with a substantial proportion of non-White ethnicity. Furthermore, a comprehensive newborn screening programme for MCADD, free at the point of use through the National Health Service (NHS), avoids potential biases related to ethnic variation in access to health care.
PATIENTS AND METHODS
Study population
Newborn screening for MCADD was introduced in the UK in 2004 as part of a pilot programme to evaluate screening test performance and examine clinical outcomes. For this study we based analyses on data from six laboratories participating in the pilot programme (Guy's Hospital, London, UK; Great Ormond Street Hospital for Children, London, UK; Royal Manchester Children's Hospital, Manchester, UK; Sheffield Children's Hospital, Sheffield, UK; Birmingham Children's Hospital, Birmingham, UK and St James' University Hospital, Leeds, UK). These laboratories provide screening for approximately half of the births in the UK. Regions served by these laboratories account for approximately 350,000 births per year and include areas of high prevalence of ethnic minorities (Figure 1). Concurrently, surveillance through the British Paediatric Surveillance Unit notified clinically presenting MCADD cases. 11 In this paper, we use data from these laboratories for the period of 1 March 2004 to 28 February 2007, during which approximately 1.1 million newborns were screened at five to ten days of age. 12
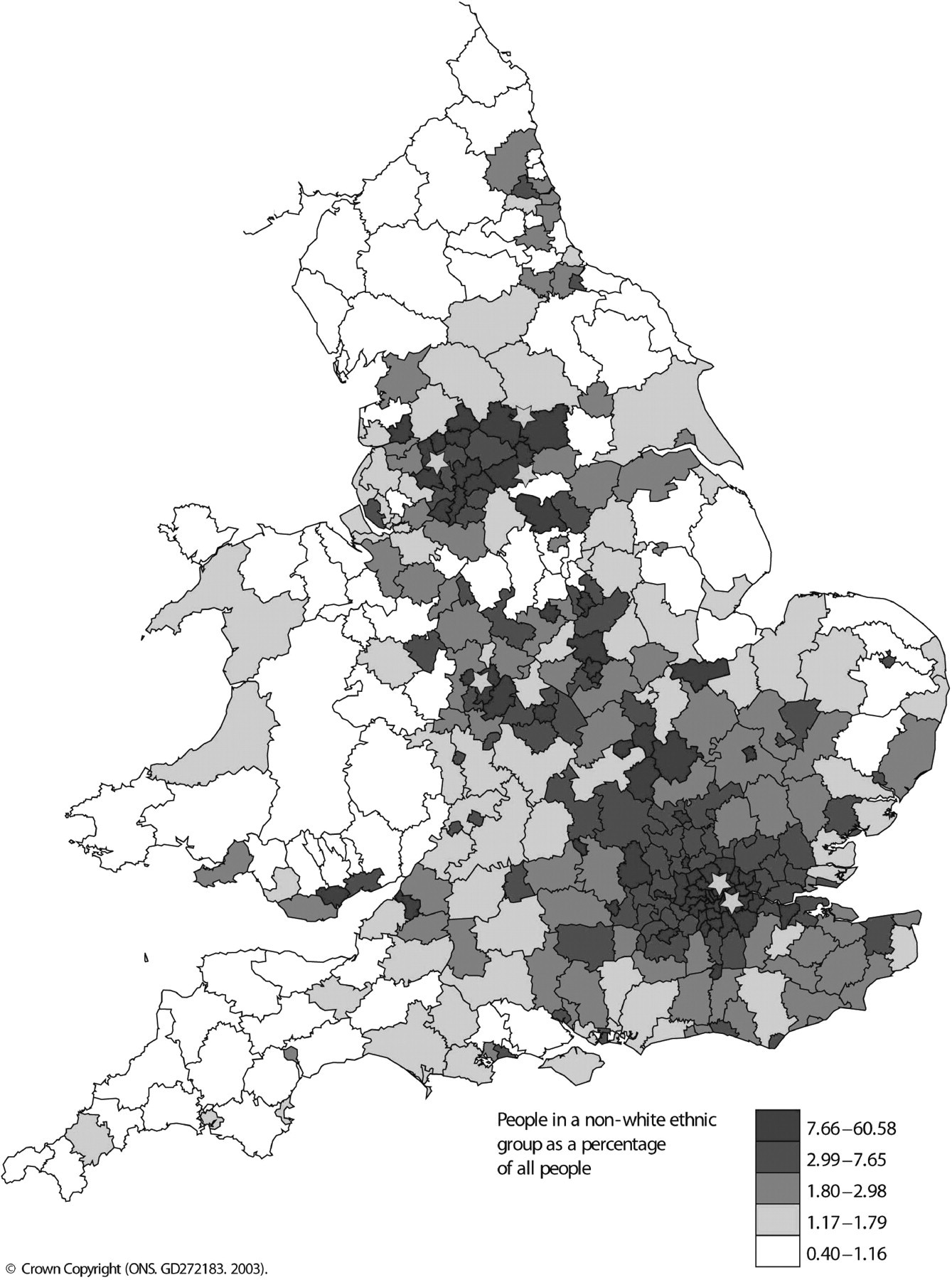
Map of England and Wales depicting the location of six newborn screening centres (shown as stars) overlaid on population density of UK residents of non-White ethnicity (2001 UK Census data). Reproduced with permission from Ordnance Survey on behalf of HMSO © Crown Copyright 2007. All rights reserved Ordnance Survey License number 0035
Case ascertainment and definition
Screening blood samples were analysed using tandem mass spectrometry (MS-MS) of underivatised blood spots using multiple reaction mode acquisitions for the quantitation of octanoylcarnitine (C8). A presumptive positive screening test for MCADD was defined by an average of triplicate C8 values greater than or equal to 0.5 µmol/L. All children with a presumptive positive test for MCADD were referred for diagnostic testing and treatment to the local metabolic specialist centre; at this point a further blood sample was obtained for repeat C8 measurement with full scan of acyl carnitines, mutation analysis and a urine sample for organic acid analysis with quantitative hexanoylglycine. Mean age (±standard deviation [SD]) at follow-up was 13 (±6) days. All children with two copies of the c.985A>G mutation had biochemical evidence of MCADD (repeat C8 ≥ 0.5 µmol/L and/or abnormal urine organic acids) and were assigned a diagnosis of c.985A>G homozygous MCADD. 13
Ethnic group
Ethnicity was used as a proxy for ancestry as information on the ancestry of the contemporary UK population, including screened newborns, is not readily available. In the UK, ethnic monitoring is mandatory at the point of access for hospital care in the NHS and each parent is required to assign child's ethnicity when the child is registered for health care. The ethnicity of children with a confirmed diagnosis of c.985A>G homozygous MCADD was obtained from clinicians' reports using UK Census categories 14 and verified by hospital records. Reported ethnicity was compared with that obtained through hospital records and showed good agreement (data not shown) when validated against parental reports of ethnicity on hospital administration database systems. For the purpose of this study, UK Census categories were combined into one of the four groups – White, Black, Asian, mixed/other – as reported in Box 1. It is important to note that in the UK Census, Asian refers to people with ancestry from the Indian subcontinent as opposed to people of Chinese, Vietnamese, Korean, Japanese or Pacific Island ancestry as is convention in the US. Peoples of East Asian ancestry were placed in the ‘mixed/other’ group and in the UK population represent approximately 13% of the ‘mixed/other’ group.
Combined 2001 UK Census categories used in MCADD screening study
White (white British, white Irish or any other white background) Black (black British, Caribbean, African or any other black background) Asian* (Asian British, Indian, Pakistani, Bangladeshi or any other background from the Indian subcontinent) Mixed/other (Mixed: White and Black Caribbean; White and Black African; White and Asian; other mixed; other: Chinese or other ethnic group)
Asian in the UK Census refers to people with ancestry from the Indian subcontinent as opposed to people of Chinese, Vietnamese, Korean, Japanese or Pacific Island ancestry as is convention in the US
Population denominators
Population denominators were obtained using 2001 UK Census estimates of total births in the UK from routine vital statistics and ethnic group and birth data of children under one year, a method originally described by Hickman et al. 15 and subsequently used by Hardelid et al. 16 to report ethnic variation in the prevalence of sickle cell disorders and phenylketonuria, respectively. In the UK Census, parents are required to assign an ethnic group for their children using a choice of categories. For the purpose of this study, we assigned children to one of four main ethnic groups by combining the 2001 UK Census categories.
UK Census population estimates by ethnic category and local area Primary Care Trust (PCT) are available on the Office for National Statistics website. 17 PCTs covering all parts of England receive budgets from the UK Department of Health and are responsible for delivering health care to their local population. PCTs served by the six newborn screening laboratories were identified from data supplied by the laboratories to the UK National Screening Committee. 18 The number of births in each ethnic group was calculated assuming that the Census data on ethnicity of children aged under one year in any given year approximates to the births for that year. The estimated proportion of births by ethnic group was then multiplied by the corresponding number of births in the six screened areas to obtain the total number of births for each ethnic group for the period 2004–2007. The denominator populations were obtained by assuming the same proportion of births by ethnic group for the years 2004–2007 as the year 2001. From this, we estimated 79.5% of children aged less than one year were White, 10.2% Asian, 5.5% mixed/other and 4.7% Black.
Sensitivity analyses were conducted for changes in the denominator population to reflect possible changes in the population distribution of ethnic groups since 2001 by increasing and decreasing the relative proportion of non-White and White births, respectively, and also for prevalence estimates in Black and Asian ethnic groups by assuming any patients of unknown ethnic background were either Black or Asian, respectively.
Prevalence rates were reported with 95% CI calculated using the exact binomial method; 19 all calculations were performed in STATA SE 9.0 (Copyright 1984–2006 StataCorp, Texas, USA). Where the estimated prevalence was zero, the upper 95% CI for the observed zero prevalence rates were calculated using a Taylor expansion to solve Hanley and Lippman-Hand's probability algorithm implying that where the upper limit of the CI for prevalence an event conditional on observing zero events in a large sample of size (n) is very close to 3/n. 20
RESULTS
In our study, we identified 64 infants (34 [53%] male) with c.985A>G homozygous MCADD, of whom 60 (93.7%) were White. No Black or Asian child with the c.985A>G homozygous mutation was diagnosed (Figure 2). Two newborns with c.985A>G MCADD were of mixed/other ethnicity and the ethnic origin of two newborns was unknown. All were diagnosed through investigation of children within the screened cohort. No false-negative homozygous c.985A>G MCADD cases were detected.
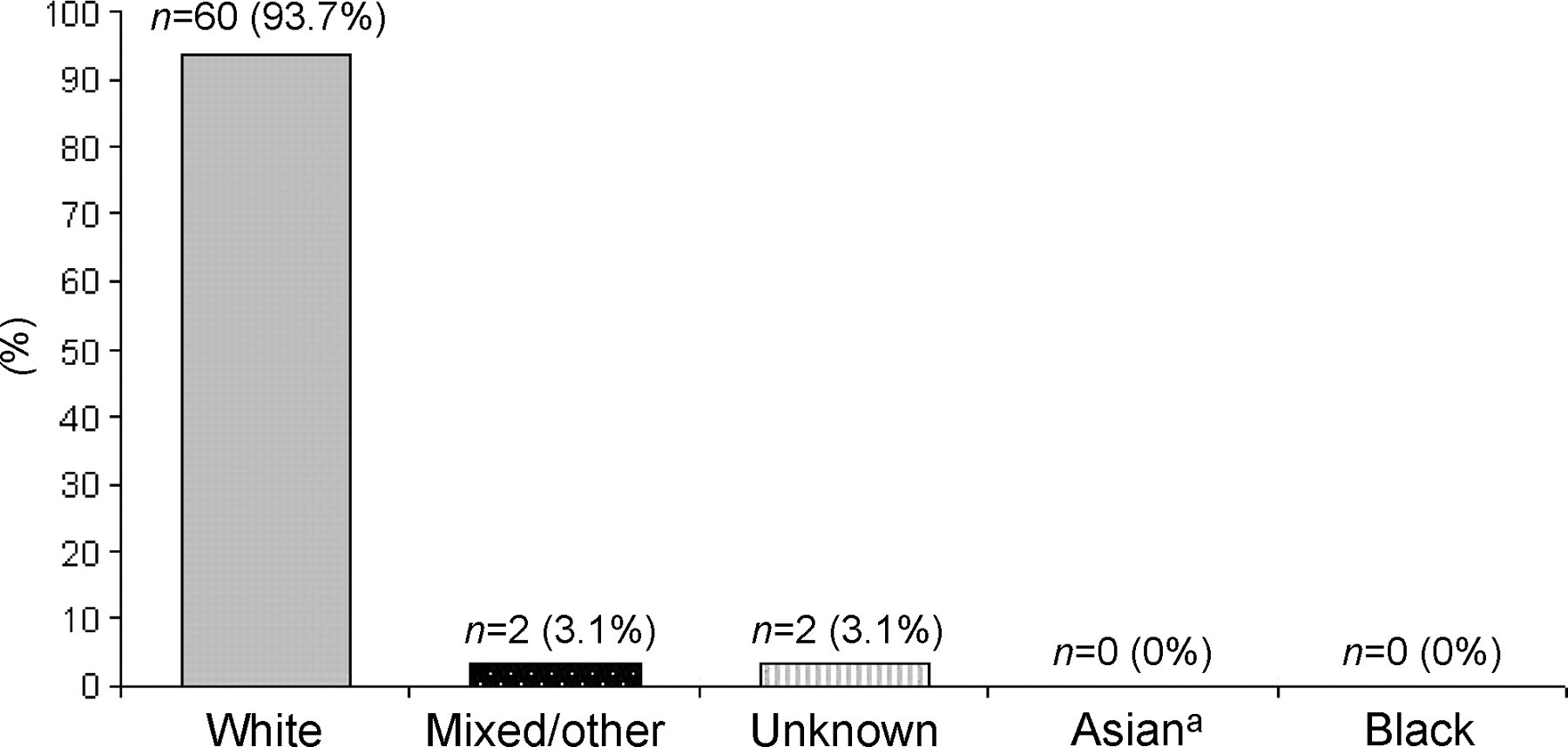
Percentage of children diagnosed with homozygous c.985A>G MCADD in six screening centres across England between 2004 and 2007 by ethnic group. aAsian in the UK Census refers to people with ancestry from the Indian subcontinent as opposed to people of Chinese, Vietnamese, Korean, Japanese or Pacific Island ancestry as is convention in the US
Children of white ethnicity represented 79.5% of the total population studied (Table 1). Estimates of the birth prevalence of homozygous c.985A>G MCADD by ethnic group are shown in Table 1.
Estimated population denominators, and birth prevalence per 100,000 by ethnic group based on 2001 UK Census data in six English screening centres, 2004 – 2007
*Asian in the UK Census refers to people with ancestry from the Indian subcontinent as opposed to people of Chinese, Vietnamese, Korean, Japanese or Pacific Island ancestry as is convention in the US
The prevalence at birth of homozygous c.985A>G MCADD was highest in the White ethnic group at 6.9 per 100,000 (95% CI 5.2–8.8). An estimated total of 163,900 Black and Asian newborns were screened; none were diagnosed with homozygous c.985A>G MCADD although had the rate been similar to that observed in the White ethnic group, 13 cases would have been expected. There was no overlap between the confidence intervals for prevalence in the Asian and White ethnic groups; however, there was some overlap with the MCADD prevalence of the children of Black and mixed/other ethnic origin. The prevalence estimates and 95% CIs for Black and Asian ethnic groups became 3.9 (95% CI 0.5–14.0) per 100,000 and 1.8 (95% CI 0.2–6.4) per 100,000, respectively, by assuming that the two children for whom ethnic origin was unknown were either both Black or both Asian.
Since there may have been a change in the proportion of births in each of the ethnic groups examined since 2001, we varied the proportion of births by ethnic groups and recalculated homozygous c.985A>G MCADD birth prevalence. For example, a 15% decrease in the proportion of births by White ethnic group yielded prevalence rates of 8.1 (95% CI 6.2–10.4) per 100,000 and a 15% increase in other ethnic groups yielded prevalence rates of 3.0 (95% CI 0.4–10.9) per 100,000, 0 (95% CI 0.2–4.2) per 100,000 and 0 (95% CI 0.5–2.7) per 100,000 for mixed/other, Asian and Black ethnic groups, respectively. The 15% change in population estimates did not change prevalence estimates significantly.
The c.985A>G carrier frequency in the White ethnic group was estimated to be 1/65 (95% CI 1/74, 1/61) assuming Hardy–Weinberg conditions.
DISCUSSION
We have used screening data from approximately 1.1 million infants born in areas of high ethnic minority prevalence to estimate the ethnic-specific birth prevalence of homozygous c.985A>G MCADD. We found this to be 6.9 per 100,000 (95% CI 5.2–8.8) in the White ethnic group, but did not find a single case of homozygous c.985A>G MCADD among children of Black and Asian ethnic origin who were screened for MCADD at birth in the UK. Furthermore, as the 95% CIs of the estimated MCADD prevalence for the White ethnic group did not overlap with that for the Asian ethnic group in our base-case analysis, we can be confident that this represents a true difference in prevalence and has not occurred by chance.
The carrier frequency for the White ethnic group as estimated from our study is comparable with that reported by Seddon et al., 10 who conducted a study in areas of the West Midlands (including Herefordshire, Shropshire and North and South Worcestershire) and Derbyshire, Leicestershire, Lincolnshire, Nottinghamshire, Rutland, South Humberside and South Yorkshire; areas which cover populations that are predominantly of white ethnicity.
Strengths of our study include the completeness of ascertainment of cases in the regions and time period of interest, the high sensitivity of MCADD screening, 21 and the large sample size on which our estimates are based. Complete ascertainment is likely as the UK's screening programme is free and universal, has a high coverage 22 and includes all ethnic groups in the regions tested. Screening programmes offered in other developed countries where access to health care is determined by the ability to pay may potentially overlook disadvantaged, minority ethnic groups. 23
Since the number of births by ethnic group is not recorded in routine health service data sources in the UK, it was necessary to estimate this from Census data to calculate the denominator population. We consider this to be the most rigorous method for estimating births by ethnic group. Moreover, large differences in the denominator have little impact on prevalence estimates for a rare condition such as MCADD.
Assessing ethnicity is not without problems as some studies have shown that up to 4.3% of category-based self-reported ethnicity questions can be missing or erroneous upon re-review. 24 However, in our study we were able to compare clinical report of ethnicity obtained through surveillance and the hospital record of ethnicity obtained form parental report at registration in one centre and found good agreement between these two sources (data not shown).
We used the Hardy–Weinberg principle to estimate the frequency of the c.985A>G allele among those of White ethnicity. Calculations were based on assumptions of a large population limiting genetic drift, absence of active natural selection and assortative mating, and served as estimates of gene proportions in our population. We found that the carrier frequency obtained was in agreement with the previously published UK findings where the study sample contained a relatively higher proportion of children of White ethnic origin. 10
Our results concur with previous reports that homozygous c.985A>G MCADD is not found in non-White populations. 25–28 Previous reports of mutation analyses based on dried blood spots on filter paper cards obtained from newborn screening programmes identified carriers of the c.985A>G mutation in Britain, Australia and North America, but not in Japan, 26,27 although the latter study was based only on 500 infants. Further investigation of the North American and Australian patients determined the patients to be of European ancestry. Moreover, the frequency of homozygous c.985A>G MCADD in Japan was estimated to be only less than one in 1,000,000 based on screening for the common c.985A>G mutation. Subsequently, a larger pilot study of expanded newborn screening which included other genotypes led to the identification of two Japanese affected individuals among 102,200 newborns screened. 29
Our results further support the suggestion that c.985A>G homozygous MCADD is a disease of White ethnic origin and the notion that it is subject to a North-western European founder effect as proposed by Gregersen et al. 7 The European distribution of c.985A>G homozygous MCADD has been reported as high in the area stretching from Russia to Bulgaria, and in all northern countries in western and middle Europe. 27 This reported distribution pattern in combination with the finding that all c.985A>G alleles belong to a single haplotype 27 suggests that the mutation occurred from a single ancestral source in the Neolithic or later period. It has been suggested, based on the differences of geographical distribution, that it was brought to Europe from the region encompassing southern Ukraine and southern Russia. 7,27
To date, the complete spectrum of ACADM mutations remains unknown, although more than 40 mutations have been identified. 30 These mutations may be of particular interest when screening non-White ethnic groups, but are also present in a significant proportion of disease-causing alleles in White populations. 31,32 Further work on characterizing mutations in other ethnic groups is currently being undertaken as screening for MCADD becomes increasingly widespread. Additional evaluation of the mutations found in MCADD newborn screening programmes serving multiethnic populations is needed and may have implications for the mutations screened for in these groups. Furthermore, greater knowledge of disease-causing phenotypes may facilitate resolution of a definitive diagnosis in patients with currently ambiguous metabolite profiles that are suggestive of MCADD as is discussed by Ensenauer et al. 25 This is likely to be of importance in delivering newborn screening programmes serving non-White ethnic groups such as in the US where confirmatory testing with MCAD gene mutation analysis is currently recommended. 33
Funding
The UK Collaborative Study of Newborn Screening for MCADD was funded by a grant from the Department of Health (England) and commissioned by the UK National Screening Committee.
Footnotes
ACKNOWLEDGEMENTS
This work was undertaken at GOSH/UCL Institute of Child Health which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. The Centre for Paediatric Epidemiology and Biostatistics also benefits from funding support from the Medical Research Council (MRC) in its capacity as the MRC Centre of Epidemiology for Child Health. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health. The authors also wish to extend their thanks to Pia Hardelid, José Leal and Dr Ying Foo for helpful advice and to Sir Muir Gray for his support in obtaining funding for this study.