
Abstract
Objectives
This paper reports early screening results from the newborn sickle cell disease screening programme recently implemented in England.
Setting England.
Screening is offered at 5-8 days of age as part of the existing bloodspot test and offered to all babies irrespective of ethnicity.
Methods
The laboratory methods recommended are high performance liquid chromatography (HPLC) and iso-electric focusing (IEF). 15 Two methods of analysis must be applied to all screen positive results. The conditions screened for are:- Sickle cell anaemia (Hb SS), Hb SC disease, Hb S/β-thalassaemia, Hb S/DPunjab, Hb S/OArab, Hb S/HPFH. Carriers identified for the common haemoglobin variants are reported to parents and follow-up counselling is offered. A bespoke laboratory quality assurance programme has been established which has defined standards of satifactory performance.
Results
Provisional figures from the first seven months of screening (up to March 2004) 108,255 infants were screened gave a screen positive rate of 1:900 for these high prevalence areas and a carrier rate of 2.7%. Figures for 2004-2005 show about 250 significant screen positive results for sickle cell disorders and about 6,500 carriers were identified. The birth prevalence for screen positive results from 2004-05 is 1:1500. We estimate that when there is countrywide data, the national birth prevalence will be about 1:2000-1:2,500.
Conclusion
The results from the national newborn sickle cell screening programme in England - show that the sickle cell disorders are as common as cystic fibrosis (CF) in England, although the distribution of cases is concentrated in London and other urban areas. The findings and approach to implementation adopted in England may be of interest to other Western European countries with increasing rates of sickle cell disease who are considering such programmes and also to other developed countries.
Introduction
It has been known for 20 years that early identification of affected infants, prescribing of penicillin and education of parents about signs and symptoms leads to reduced infant mortality. Hence, two systematic reviews of screening for haemoglobinopathies both recommended implementation of newborn screening for sickle cell disorders for England.6,7 Following this a commitment was made in 2000 by the UK government to implement ‘effective and appropriate screening programmes for women and children including a new national-linked antenatal and neonatal screening programme for haemoglobinopathy and sickle cell disease’. 8 The National Health Service (NHS) Sickle Cell and Thalassaemia Screening Programme was set-up to implement appropriate newborn and antenatal screening throughout England. This paper reports early screening results from the newborn programme, the first part of the programme to be implemented nationally.
Methods
Programme policy and guidance
The policy for newborns supported by the National Screening Committee is to offer screening to all babies in England irrespective of ethnicity as part of the existing bloodspot screening arrangements as the only practical and robust implementation solution. 9 This was despite alternative proposals in the two systematic reviews.6,7 The decision to adopt this approach was made based on difficulties and problems reported by the USA and France on the experience of targeted programmes. It favoured equity over cost-effectiveness and also considered the practical issues such as how the programme is offered.9–11 The overall aim of the national newborn programme is to identify infants at risk of sickle cell disorders to allow early detection and to minimize deaths and complications through early treatment and care.12,13
Screening is offered at 5-8 days of age as part of the existing newborn dried bloodspot screening programme. The laboratory service in England consists of 13 centralized biochemistry newborn screening laboratories. These laboratories routinely perform newborn screening for phenylketonuria, congenital hypothyroidism and in some areas cystic fibrosis and in six laboratories medium chain acyl-CoA dehydrogenase (both currently being introduced nationally).
The programme specifies the conditions to be screened for and for which early intervention is likely to be beneficial. 14 These are: sickle cell anaemia (Hb SS), Hb SC disease, Hb S/β-thalassaemia, Hb S/DPunjab, Hb S/OArab and Hb S/HPFH. Other clinically significant haemoglobinopathies likely to be detected by the programme are β-thalassaemia major, β-thalassaemia intermedia, Hb-H disease, Hb-E/β-thalassaemia and Hb-SE. Clinical follow-up is recommended for these conditions although they do not meet the criteria for screening and are not the main focus of the programme. The methods recommended do not reliably detect all thalassaemia conditions and never detect beta-thalassaemia carriers.
The haemoglobin make-up of newborns is different to adults and different methods are needed for screening. Normal newborns have predominately fetal haemoglobin and a variable amount of Hb A. The laboratory methods recommended by the Programme for newborn screening and diagnosis are high performance liquid chromatography (HPLC) and iso-electric focusing (IEF). 15 The Programme requires two methods of analysis be applied to all screen-positive results obtained from dried bloodspot samples. Either of the methods can be used for first-line screening, the alternative procedure being used for second-line testing to validate the presumed identity of the haemoglobin variant and ensure high specificity. Hb D-Punjab has not been characterized at the mutation level in this screening programme. However, D-Punjab has a characteristic appearance on HPLC, which distinguishes it form D-Iran and D-Korle Bu, (the next two most frequent of the Hb D's). Furthermore, the addition of IEF as a second test gives extra certainty to the classification. Nevertheless, it must be emphasized that these are screening results, and not diagnostic certainties. HPLC is used as the first-line method in all except three laboratories. The second method is sometimes undertaken by a separate laboratory with wide haematological experience. A national framework agreement has been established by the NHS Purchasing and Supplies Agency making the prices clear, to avoid repeated tendering and try and use this leverage to improve the quality of the technology available.
Carriers identified for the common haemoglobin variants (S,C, D and E) are reported to parents and follow-up counselling is offered to ensure understanding of the difference between carrier state and disease and also the genetic significance of the condition. This is in-line with recent Human Genetics commission guidance ‘Whilst some would say this information ought not to be divulged, others, including patient representatives, argue strongly that the result of the tests, once generated, should be given to the parents. We support this latter position and this information should not be withheld from parents who indicate that they wish to have it when agreeing to have their child tested.’ (Making Babies 2006)
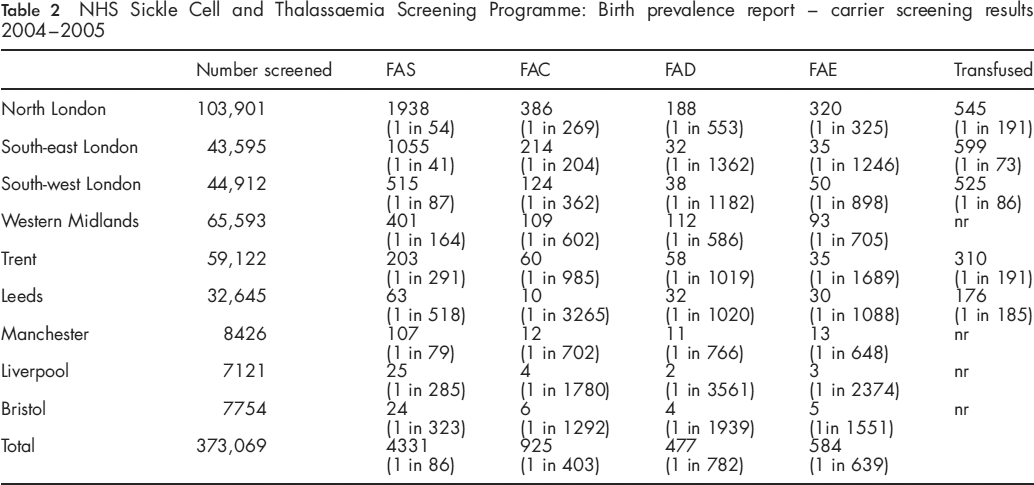
As a linked newborn and antenatal programme consistency in reporting by and between the antenatal and newborn programmes is important to ensure that accurate information is provided to those who have screen-positive results. About half of all carrier infants, identified by newborn screening, will have carrier mothers who should (assuming antenatal screening is in place) be identified by the antenatal programme. Linking this information allows those who counsel the parents to distinguish straightforward cases where a newborn carrier result is ‘expected’ (because the mother is already known to be a carrier) from those whom it is ‘not expected’ as the mother is not carrier. In these latter cases, information about the biological father could reveal non-paternity if counselling is not handled sensitively. Currently, the information to access information on mothers screening record and screening results when a baby is identified as a carrier remains difficult and time consuming and is not always possible in routine practice. A variant carrier identified in the newborn period should also alert health professionals to the fact that the parents could be an ‘at-risk’ couple for a future pregnancy and the parents should be advised about this risk if it has not already been identified. It is important to note, and to communicate with parents where relevant, that unlike antenatal screening, newborn screening does not identify thalassaemia carriers although where the mother is a carrier parents may commonly request this information on their infant.
Implementation
The main phase of implementation started in September 2003 in areas of higher prevalence where some screening was already taking place in three laboratories covering about 15% of the country mostly in London. It was then rolled out to lower prevalence areas over three phases and completed in July 2006 covering approximately 600,000 babies born annually. Funding of £4 per baby for laboratory staff and reagent costs and equipment was provided to start the programme and some funding for additional counselling was also provided with the NHS expected to pick up running costs subsequently. A national framework agreement provided standard equipment rental costs for both screening methods. Formalization of clinical networks and arrangements for quality assurance and audit of the programme are still under development. As part of implementation a bespoke laboratory quality assurance programme has been established by UK NEQAS and all the laboratories take part in the National quality assurance scheme which has defined standards of satisfactory performance.
To achieve the potential benefits of screening it is essential that affected infants are reliably diagnosed, reports are issued and that co-ordinated clinical follow-up for all parts of the country are established. Experience from the USA showed wide geographic variation in mortality attributed to variation in clinical follow-up arrangements and in the UK before screening, problems with care were demonstrated.15,16 Experience from the Jamaican cohort showed improved survival in the later part of the cohort due to provision of effective follow-up and care. 17 Therefore, the programme is now working, alongside commissioners and clinicians, to formalize a national network of clinical specialist centres to ensure adequate failsafe arrangements for follow-up, clinical care and quality assurance are established in all parts of the country.12–18
Results
Provisional figures from the first seven months of screening (up to March 2004) 108,255 infants were screened in London and the Midlands. There were 125 clinically significant screen-positive results identified of which 110 were in London, in addition 2950 carriers were identified. Of the screen-positive results 86 were reported as having fetal haemoglobin and haemoglobin S (reported a FS and likely to be sickle cell disease), 31 were reported as having fetal haemoglobin (F), haemoglobin S (S) and haemoglobin C (C), reported as FSC and likely to represent HbSC diseases, 31 were reported as having fetal haemoglobin, and haemoglobins S and C (reported as FSC and likely to be HBSC disease, and seven were reported as FS-Other (meaning having fetal haemoglobin with S and another less usual haemoglobin such as D or E). There were also nine infants with only Hb F (suggestive but not conclusive of beta thalassaemia major). This is a screen-positive rate of 1:900 for these high prevalence areas and a carrier rate of 2.7%.
NHS sickle cell and thalassaemia screening programme: birth prevalence disease summary results 2004-2005
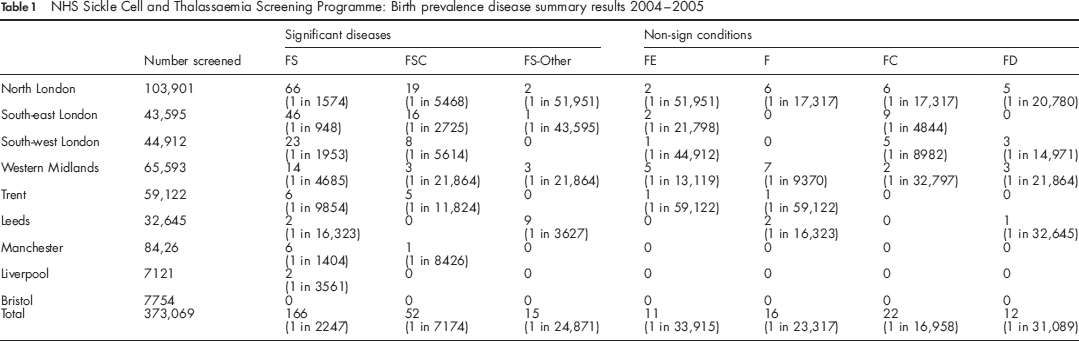
Although there is no systematic check of all normal results to detect missed cases, it is possible to check the neonatal screening result on any baby subsequently diagnosed. So far all non-predicted sickle disease children have been movements into the screening areas and not failures of the screening methods.
NHS sickle cell and thalassaemia screening programme: birth prevalence report - carrier screening results 2004-2005
Discussion
This is the first report of findings and results from the national newborn sickle cell screening programme in England - the first country in Western Europe to introduce such a country-wide screening programme. The results show that the sickle cell disorders are as common as cystic fibrosis in England, although the distribution of cases is concentrated in London where it is about five times as common as cystic fibrosis. Given current trends in population migration into the country these conditions are likely to become even more common over the next decade. They support the case for the urgent need to develop services to care for the significant number of affected patients4,5 in all parts of the country. These figures show a higher birth prevalence than previous estimates.6,7
Limitations of the study in terms of the accuracy of the prevalence figures reported are firstly that it reports screening and not confirmatory diagnostic results and therefore the final results are likely to be slightly lower than these figures. The actual prevalence of the sickling conditions are likely to be reduced by about 2% when cases of S-HPFH are eliminated at the diagnostic stage. (Joan Henthorn, personal communication). Secondly, there may be a few cases missed due to incomplete coverage of the ‘guthrie’ programme in groups at highest risk. 19 The results exclude babies who move into the country after birth. Reports from several centres are that an almost equal number of children seen in the clinic are arrivals after birth (A Will in Manchester and D Rees personal communication). Some infants may not have been identified as a result of transfusions before the collection of the bloodspot samples (which invalidates the screening result) and a lack of follow-up or incomplete coverage of the bloodspot programme, but these figures are unlikely to contribute more than 1-2% to the total (based on reports from the screening laboratories). Overall, these limitations do not change the main findings of this report, which shows that sickle cell disease is an increasingly important public health issue in England. The condition is unequally distributed across the country; affected infants are mostly located in the more disadvantaged parts of major cities. Without effective follow-up care deaths from this condition may contribute to inequalities in infant mortality.
The findings and approach to implementation adopted in England may be of interest to other Western European countries with increasing rates of sickle cell disease who are considering such programmes and also to other developed countries. 20 The consistent national policy and rapid implementation of the programme contrast with the USA where policy and implementation has been done on a state-by-state basis taking over 20 years to cover the whole country. The challenge for the NHS in England now is the establishment of an effect whole country managed network of clinical services, and the monitoring the clinical outcomes of these services and the screening programme, to support the clinical needs identified by this screening programme and to ensure that the potential benefits of the programme are achieved.
Funding
Department of Health central funding to support implementation including programme Centre costs.
Footnotes
Acknowledgements
Screening laboratories for reporting results. Many committed professionals who made their own contribution and supported the implementation the programme making it possible to implement in under three years. Two anonymous referees for helpful comments.