
Abstract
Background
Catecholamine-producing neuroendocrine tumours are found in chromaffin cells of the adrenal medulla (phaeochromocytoma) or extra-adrenal paraganglia (paraganglioma), known collectively as PPGLs. In approximately a quarter or more of cases of PPGL, these rare tumours arise as a result of germline mutations of several tumour susceptibility genes. At the Crosshouse laboratory, urine tests include free metadrenalines (fMAs) (also known as free metanephrines) which demonstrate superior sensitivity over that obtained by urinary vanillyl mandelic acid, catecholamines or plasma catecholamines in the diagnosis of PPGL. This retrospective audit was to determine if urinary fMAs offered discrimination among the hereditary forms of PPGL.
Methods
Retrospective biochemical and genetic data were gathered from 1997 to 2011. The identified urine specimens were those obtained at the time of first diagnosis or recurrence of PPGL. Results of catecholamines and metabolites were standardized as multiples of their respective relevant upper reference limits (URLs).
Results
Results were available for 29 affected patients (15 females and 14 males), median age 26 (range 9–63) years, comprising three mutation groups: succinate dehydrogenase subunit B or D ([SDHB/D] 16 patients), multiple endocrine neoplasia type 2 ([MEN 2] 6 patients) and von Hippel–Lindau disease ([VHL] 7 patients). The parent catecholamines exhibited increased values for noradrenaline (NA) and/or adrenaline (AD) for 25/29 (86.2%) patients. Either or both free normetadrenaline (fNMA) and fMA were elevated in 29/29 (100%) patients.
Conclusions
The ratio of the multiples of URL for fMA/fNMA displayed a clearer separation of MEN 2 patients from those with SDHB/D or VHL than did the equivalent AD/NA ratio.
Introduction
Catecholamine-producing neuroendocrine tumours are found in chromaffin cells of the adrenal medulla (phaeochromocytoma) or extra-adrenal paraganglia (paraganglioma) and known collectively as PPGL. 1 Although most cases appear to arise sporadically, hereditary forms of PPGL are associated with several syndromes, most notably: multiple endocrine neoplasia type 2 (MEN 2), von Hippel–Lindau disease (VHL), neurofibromatosis type 1 (NF 1) and familial PPGL resulting from mutations in genes that encode subunits of the succinate dehydrogenase complex, most commonly succinate dehydrogenase subunit B (SDHB) and succinate dehydrogenase subunit D (SDHD). 2 Hereditary forms of PPGL tend to be diagnosed earlier in life than sporadic forms, often before age 40 y. 1,2 The occurrence of PPGL is rare in children. However, when found in childhood, it can often be extra-adrenal and associated with hereditary syndromes. 3
Following the discovery by Julius Axelrod that methylated forms of the parent catecholamines were major metabolic end products of noradrenaline (NA) and adrenaline (AD), it was noted that these metadrenalines (also known as metanephrines) were mainly present in human urine as conjugates. 4 In order to improve the assay of metadrenalines in urine, for example by Pisano's method which entails oxidation to vanillin followed by spectrophotometry, it was deemed necessary that a first step was to convert these conjugates to their free forms by acid hydrolysis, thereby greatly increasing the resulting analytical signal. 5,6 Therefore, unlike other parameters of catecholamine excretion in urine (e.g. NA, AD, vanillyl mandelic acid [VMA] or homovanillic acid [HVA]), the assay of metadrenalines has historically been preceded by a deconjugation step. With the advent of improvements in analytical techniques which allowed measurement of normetadrenaline and metadrenaline separately, for example by high-performance liquid chromatography (HPLC), this deconjugation procedure has been continued, is unique to assays of metadrenalines, and is described in the literature as ‘urinary fractionated metadrenalines or metanephrines’. 1 Among the readily available urine tests for detection of PPGL, fractionated metadrenalines exhibit superior clinical sensitivity when compared with other procedures such as urine catecholamines or VMA. 1,7 This is due to the continuous production of unconjugated O-methylated metabolites within tumours from catecholamines leaking from chromaffin stores. 8 However, despite the superior sensitivity of urinary fractionated metadrenalines, this test exhibits relatively poor specificity with a concomitant much higher frequency of false-positive results. 1,7 The metadrenalines are present in urine mainly as sulphate conjugates which are formed by the actions of a specific sulphotransferase found in the gastrointestinal tract. 4,9 A recent comprehensive study from Holland and Germany has shown that consumption of catecholamine-rich food products results in sustained and substantial increases in urinary conjugated normetadrenaline but negligible effects on free normetadrenaline (fNMA) and metadrenaline (fMA), a fact which may explain the high false-positive rate and low specificity associated with assays which employ a preliminary deconjugation step. 10 Interestingly, a recent evaluation of the diagnostic accuracy of urinary fMAs for the identification of PPGL conducted in our tertiary care centre has demonstrated superior sensitivity over that obtained by, for example urinary VMA or catecholamines. 11 In addition, the specificity achieved for urinary fMAs in our study was 94%, 11 which is significantly more favourable than that reported for urinary fractionated metadrenalines of only 64–69%. 1,7
Mutation testing is routinely available for four genes (RET, VHL, SDHB, SDHD) associated with MEN 2, VHL syndrome and familial PPGL syndromes, respectively. At the first international symposium on phaeochromocytoma, it was estimated that approximately a quarter or more of PPGL cases, without an obvious syndrome or family history, do result from otherwise unsuspected mutations in one of these four genes, and indicates a need for more widespread genetic testing of patients with these tumours than is currently practiced. 12 However, it was considered that it was neither appropriate nor cost-effective to test for every disease-causing gene in every patient manifesting PPGL, and consideration of tumour location, presence of multiple tumours, presence of metastases, age at presentation and type of catecholamine produced would be useful in deciding which genes to test. 12
As regards the biochemical phenotype, it has been noted that phaeochromocytomas in patients with MEN 2 always produce adrenaline, unlike, for example, those with VHL syndrome. 12 In a recent, large, multicentre, retrospective analysis of data from 173 patients with hereditary PPGLs (VHL, MEN 2, NF 1, SDHB and SDHD) conducted by Professor Eisenhofer and his colleagues, it was observed that both plasma fMAs and urinary fractionated metadrenalines were considerably more effective in distinguishing NF 1 and MEN 2 patients from those with mutations of either VHL, SDHB or SDHD genes than measurement of the parent catecholamines in plasma or urine. Hence, it was established that measurements of the O-methylated metabolites of catecholamines, in addition to providing useful biomarkers for diagnosis of PPGLs, also enable discrimination of different hereditary forms of these tumours and thereby guide more cost-effective genetic testing. 13 However, although comprehensive, this study did not include measurement of the urinary fMAs. Therefore, the purpose of the present laboratory-based, retrospective study was to determine whether the urinary fMAs were likely to offer as good discrimination among the hereditary forms of PPGL as these other two analytical approaches.
Patients and methods
Close links exist between the West of Scotland Regional Genetics Service and the Endocrinology Department at the Western Infirmary Glasgow. This is a tertiary referral centre serving a population of approximately 2.5 million. On the basis of local guidelines, those affected with PPGL who are either under the age of 50 y or manifest malignant or multifocal disease, or have a positive family history, are offered genetic testing. Testing is also made available to other family members. Analysis of SDHB, D, VHL and RET, by complete gene sequencing, is carried out by the Scottish Molecular Genetics Consortium laboratory in Dundee. Our local genetics database is maintained by the West of Scotland Regional Genetics Service. Inspection of this genetic database allowed identification of patients exhibiting hereditary forms of catecholamine-secreting PPGL. The present laboratory-based study consisted of gathering biochemical data obtained between 1997 and 2011 from among only those patients who had been tested for urinary catecholamines and metabolites at the Crosshouse laboratory and identified as exhibiting hereditary forms of catecholamine-secreting PPGL. All of the data were obtained under conditions of regular clinical care and during clinicians’ own investigative procedures. In each case, the identified urine specimens were those obtained at the time of first diagnosis, or at the time of recurrence of PPGL. Where more than one urine specimen was received at the time of diagnosis or recurrence, the mean results for each analyte from that patient were calculated. Results from 24-h urine specimens, collected into hydrochloric acid preservative, were available from adult patients. Among those presenting in childhood, random urines were available and were treated as described previously. 14 Urinary NA, AD, dopamine (DA), VMA, HVA, fNMA and fMA had been measured by HPLC with electrochemical detection at the Crosshouse laboratory as described previously. 15 The reference intervals for adults were NA (<900 nmol/24 h), AD (<230 nmol/24 h), DA (<3300 nmol/24 h), 16 VMA (<35 μmol/24 h), HVA (<40 μmol/24 h), 17 fNMA (<650 nmol/24 h) and fMA (<350 nmol/24 h). 15 In the case of random urines from children, results were expressed as millimole per mole creatinine, and the various age-related upper reference limits were as previously described. 14
Statistical analysis was performed using the MedCalc statistical software package (version 11.3.3.; MedCalc Software, Mariakerke, Belgium). Differences between groups was assessed by one-way analysis of variance using the non-parametric Kruskal–Wallis (H-test) and were considered to be significant if P < 0.05 and were followed by pairwise comparison of subgroups. 18
Results
Results were available for 29 patients (15 females, 14 males) of median age 26 y (range 9–63). Of these, 23 sets were obtained at the time of first diagnosis and from a further six patients, at the time of identification of recurrence. For these latter six patients (4 SDHB/D, 1 MEN 2 and 1 VHL), results of urinary fMAs at time of first diagnosis were not available at the Crosshouse laboratory. There was no significant difference in the patients’ ages at time of identification between these two groups (P = 0.63). The 29 patients were subdivided into three mutation groups comprising: (1) SDHB/D n = 16, median age (range) 28 (11–63) years; (2) MEN 2 n = 6, median age (range) 40 (11–53) years; and (3) VHL n = 7 median age (range) 20 (9–27) years.
None of the six MEN 2 individuals were related. Two of the VHL individuals were distantly related but both presented separately and a family link was identified later. Two SDHB cousins were also affected but both presented with disease at similar ages and time. The West of Scotland has a founder SDHB mutation proven by haplotype analysis. 19 Three individuals (1 SDHB, 2 VHL) had disease detected during surveillance testing after a positive predictive genetic test for a known familial mutation.
Medians and observed ranges of urinary free catecholamines and their metabolites, expressed as multiples of their upper reference limits, for each of the three mutation groups
VMA, vanillyl mandelic acid; HVA, homovanillic acid; NA, noradrenaline; AD, adrenaline; DA, dopamine; fNMA, free normetadrenaline; fMA, free metadrenaline
Proportions of patients with hereditary phaeochromocytoma/paraganglioma who exhibited abnormally elevated urinary catecholamines or free metadrenalines in each of the three mutation groups
NA, noradrenaline; AD, adrenaline; fNMA, free normetadrenaline; fMA, free metadrenaline
*Indicates one result not available due to analytical interference
It can also be seen in Table 2 that only three of six patients in the MEN 2 group exhibited elevated AD, while all six displayed increased fMA. In the SDHB/D and VHL groups, all available AD results were normal while only one fMA value was above its URL. If results are expressed as multiples of their respective URL, then in MEN 2 patients, analysis of variance indicated that the results of NA (P = 0.0044) and fNMA (P = 0.0125) tended to be significantly lower than those of patients in the SDHB/D and VHL groups. Similarly, for MEN 2 patients, analysis of variance indicated that the results for AD (P = 0.0097) and fMA (P = 0.0006) tended to be significantly higher than those in the SDHB/D and VHL groups. Therefore, an obvious combination of biochemical results to investigate, in order to better aid discrimination of MEN 2 patients from those harbouring mutations of SDHB/D or VHL, would be the ratio of the multiples of the URL values for AD/NA and fMA/fNMA, respectively. Such a multiple variables graph for each of the genetic mutation groups is given in Figure 1 and indicates that the ratio of the multiples of URL for fMA/fNMA displayed a clearer separation of MEN 2 patients from those with SDHB/D or VHL than did the equivalent AD/NA ratio.
Comparison of the ratios of upper reference limits (URLs) of urinary adrenaline to noradrenaline (AD/NA) and of ratios of URLs of urinary free metadrenaline to normetadrenaline (fMA/fNMA) in three mutation groups: multiple endocrine neoplasia type 2 (MEN2); succinate dehydrogenase subunit B or D (SDHB/D); and von Hippel–Lindau syndrome (VHL) (horizontal lines = median values)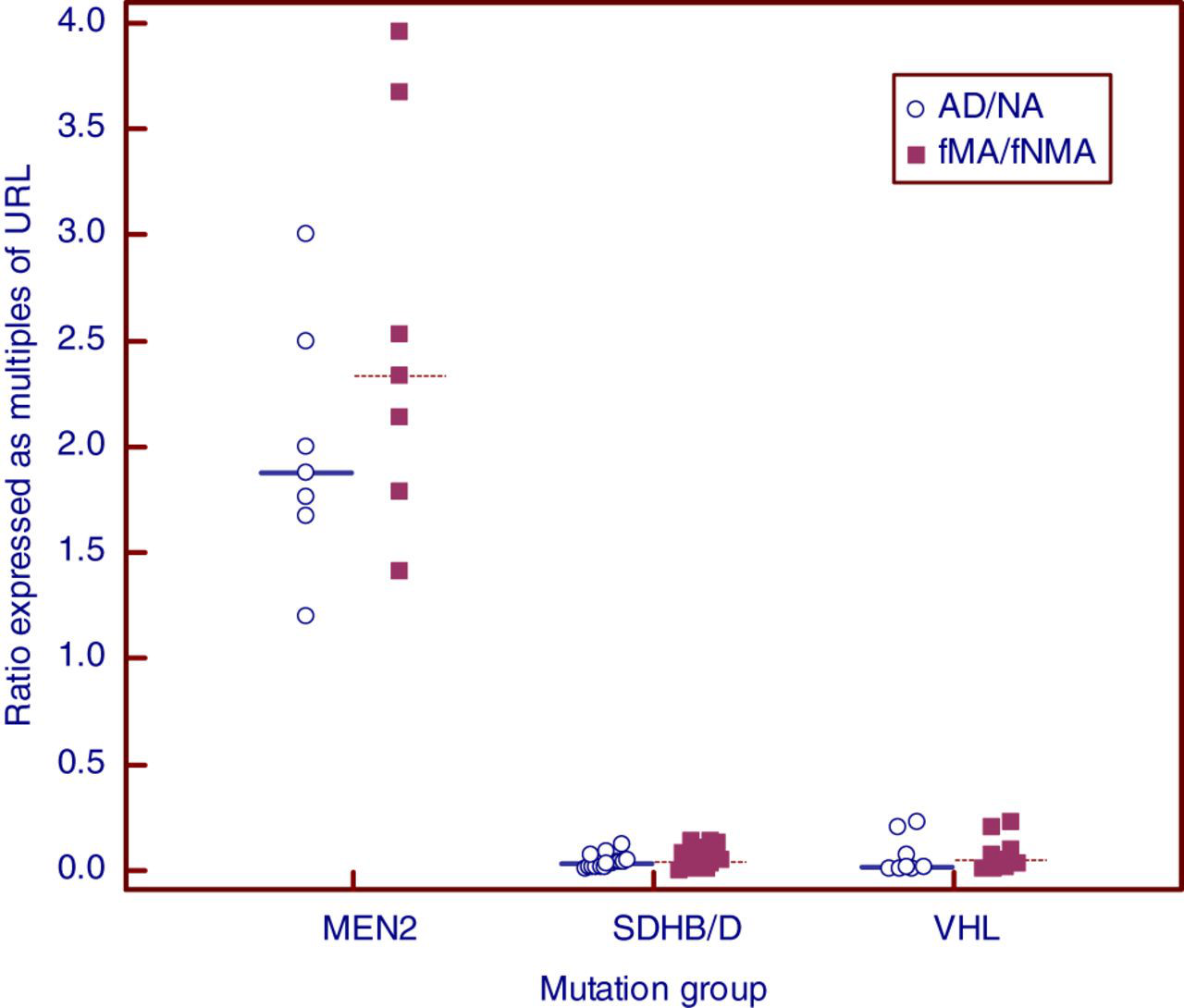
Discussion
Clearly, in addition to better detection of PPGL, measurement of the urinary fMAs appears to allow better discrimination of MEN 2 patients from those with SDHB/D or VHL than does the measurement of the parent catecholamines as illustrated in Figure 1 from this small cohort. It had been observed previously by Professor Eisenhofer et al. 20 that in MEN 2, unlike those with VHL, AD production represents a consistent finding among different kindreds and in large numbers of patients manifesting phaeochromocytoma. However, using plasma free measurements, they also noted that half of all MEN 2 patients with phaeochromocytoma had normal AD concentrations whereas all had elevated fMA. 20 This is similar to the findings in the present study using urinary free measurements (Table 2), where in MEN 2 patients, AD was within normal limits in three out of six patients, whereas fMA was abnormally elevated in every case, thereby better reflecting differences in tumour catecholamine phenotype than the parent catecholamines.
In the present study, results of the various analytes were expressed as multiples of URL. This was for two reasons: first, to better illustrate the relative differences of, for example AD over NA, or fMA over fNMA, and second, for practical reasons, since a high proportion of hereditary forms of PPGL may arise in children from whom only random urines, rather than 24-h collections, may be likely to be available. Calculation of multiples of URL using age-appropriate ranges allows relative comparison of data throughout the age ranges likely to be encountered in hereditary PPGL.
It is noteworthy that there is a wide variation in the published upper reference limits for urinary catecholamines in different laboratories, 21 and selection of unsuitable values would impact on the findings of a study such as the present one. There are several issues to be considered when attempting to establish suitable reference limits for urinary catecholamines and metabolites. One of these issues is the choice of reference population. The use of a healthy population is inappropriate and would result in an unacceptably high number of false-positives. It is more appropriate to use data from patients suspected of having a phaeochromocytoma but where the diagnosis was subsequently rejected. 22,23 Another issue to be considered is the population distribution. For example, the frequency distributions for urinary output of both NA and AD in a reference population show a positive skew 16 and an assumption of normality would also increase the number of false-positives unless it is taken into account. 23 Such data may be normalized, for example by logarithmic transformation or by employing non-parametric statistics. 15,16,22,23 The upper reference limits for urinary catecholamines and metabolites used in the present study were established taking account of both factors. In addition, we determined their practical validity by a peer-reviewed study using receiver operating characteristic curve analysis, 11 and our URL values are similar or identical to those quoted by several other laboratories employing HPLC techniques. 22,24,25
Conducting a study of this type and extracting from it meaningful information does represent a considerable challenge given that we are dealing with: (1) a relatively rare condition (PPGL); (2) an even rarer subgroup (hereditary forms of PPGL); and (3) a relatively new and uncommonly available biochemical test (urinary fMAs). Therefore, the total sample size is, inevitably, a relatively small one (n = 29) and the mutation subgroups within it are represented by small numbers of patients. As patient numbers increase with the passage of time, it might be interesting to look for the relationships between analyte combinations and patient age at presentation, 26 or variations in tumour size among the subgroups. For example, in a previous report in a cohort of 18 patients unselected for the presence of genetic mutations, we observed that only urinary fNMA + fMA correlated positively with tumour volume, whereas neither urinary NA + AD, VMA nor plasma NA + AD showed a correlation with tumour volume. 11 In addition, a further interesting comparison would be to identify a control group of patients who had exhibited catecholamine-secreting PPGL but were found to harbour none of the identified genetic susceptibility mutations following appropriate laboratory analysis.
The present study should be regarded as a novel early step in the promotion of the measurement of urinary fMAs, as a potential improvement, not only in the detection of PPGL, 11 but also in the discrimination of different hereditary forms of the condition. For example, a further challenge is the discrimination of patients with SDHB and SDHD mutations from those with VHL mutations using biochemical measurements as an additional and preliminary guide to cost-effective genotyping. As has been demonstrated recently, additional measurement of free 3-methoxytyramine in plasma enabled this discrimination in 78% of cases. 13 In recent years, the Crosshouse laboratory has added the urinary free f3MT test to its armamentarium. 14 As more cases of hereditary PPGL are identified, further work is required to determine if urinary free f3MT shows similar discriminatory power to that of plasma measurements.
DECLARATIONS