
Abstract
Investigating persistent hyperferritinaemia without apparent iron overload is challenging. Even when inflammation, cirrhosis, Still's disease, fatty liver and malignancy are excluded, there remains a group of patients with unexplained hyperferritinaemia for whom rare forms of haemochromatosis (ferroportin disease) are a consideration. Preliminary results suggest that abnormal percentage glycosylation of serum ferritin is seen in some cases of genetically determined hyperferritinaemia. Serum ferritin is normally 50–81% glycosylated, but low glycosylation (20–42%) prevails in hereditary hyperferritinaemia cataract syndrome. This contrasts with hyperglycosylation (>90%) associated with the benign hyperferritinaemia related to missense L ferritin (p.Thr30Ile) mutation. Here, we describe two novel missense L ferritin variants also associated with hyperglycosylation, p.Gln26Ile and p.Ala27Val. Ferritin glycosylation, a comparatively simple measurement, can identify patients for DNA sequencing as hyperglycosylation (>90%) is associated with benign hyperferritinaemia and mutant L ferritin chain.
Introduction
Ferritin, the principal iron storage protein, has a molecular weight of approximately 500 kDa. It is comprised of 24 subunits containing H type (heavy, 21 kDa) and L type (light, 19 kDa) polypeptide chains encoded by two different genes. Together, these subunits form a spherical protein shell capable of storing iron in its central core.
Serum or plasma ferritin measurement is widely requested as an indicator of body iron reserves, though well-documented limitations to this clinical application exist. 1,2 While a low serum ferritin concentration reliably indicates depleted iron reserves, normal or high concentrations obtained during an acute phase response require caution in their interpretation and may not reflect body iron stores. 2
Hyperferritinaemia with normal or low serum iron and transferrin saturation can be difficult to explain if coexisting inflammatory conditions, acute or chronic liver disease and malignancy are excluded, and when HFE (high iron Fe gene) genetic studies reveal a wild-type sequence.
Two genetic conditions associated with hyperferritinaemia without iron overload in otherwise healthy individuals have been described. In hereditary hyperferritinaemia cataract syndrome (HHCS), point mutations in the iron-responsive element (IRE) for L ferritin chains (FTL) result in autonomous FTL synthesis, unregulated by iron reserves. Apart from early cataract development due to formation of L ferritin crystals in the lens, affected individuals are asymptomatic. 3,4 The incidence of HHCS has been suggested to be at least 1:200,000. 5 Recently, a second form of genetic hyperferritinaemia without iron overload or cataracts has been described. 6 In this condition, there is an amino acid substitution in the FTL (isoleucine replaces threonine at position 30 in the A helix). The resulting FTL appears unusually amenable to glycosylation, leading to serum glycosylated ferritin values consistently >90%. Individuals affected by either this condition or HHCS are at risk of inappropriate venesections and invasive investigation (e.g. liver biopsy) to exclude type IV haemochromatosis (ferroportin disease). 5 The exact incidence of ferroportin disease, in which an iron transport defect leads to iron overload and raised ferritin with normal or low transferrin saturation, is unknown. There appear to be no data regarding ferritin glycosylation in ferroportin disease, but in this laboratory, a normal value (70%) was obtained for a single confirmed case.
While intracellular ferritin consists of both light (L) and heavy (H) chains in varying proportions depending on the tissue of origin, circulating ferritin is almost exclusively L chains, of which 50–81% are glycosylated. Glycosylation is thought to occur during secretion into the extracellular fluid. Where hyperferritinaemia arises due to malignancy or tissue damage with release of intracellular ferritin (e.g. liver cirrhosis, Still's disease), ferritin is predominantly unglycosylated, but for hyperferritinaemia of inflammation without tissue destruction, the proportion of glycosylated serum ferritin is normal as the usual secretory processes operate. 1,7 Worwood et al. 8 developed a procedure to measure percentage glycosylated ferritin in serum, showing how this varied in different disease conditions. These findings have not been widely utilized until recently when Kannengiesser et al. 6 demonstrated hyperglycosylation of the genetically determined variant FTL associated with hyperferritinaemia.
Here, the percentage of glycosylated ferritin was determined in patients for whom hyperferritinaemia was unexplained, as a preliminary screen to identify candidates for genetic analysis.
Methods
Glycoslyated ferritin was measured in the serum of six patients with persistent hyperferritinaemia (two having become severely anaemic following a programme of venesection), where an explanation for the hyperferritinaemia was sought by the clinician managing the patient. Other than hyperferritinaemia, none displayed clinical evidence of iron overload and in all patients transferrin saturation was within normal limits. Inflammation and liver disease had been effectively excluded.
Case 1
A man aged 45 years had originally presented with tiredness and an acute chest infection. Serum ferritin was 1618 μg/L (reference range: 22–322), serum iron was 18 μmol/L (reference range: 11–31) and transferrin saturation was 28% (reference range: <45%). Hyperferritinaemia persisted though the patient was well. C-reactive protein, erythrocyte sedimentation rate, liver function tests and glycaemic control were normal. He had no cataracts, but there was thought to be a history of iron overload on his maternal side. HFE analysis showed homozygosity at positions 282 and 63 for the normal wild-type sequence. Quantification of hepatic iron concentration by quantitative magnetic resonance imaging scanning (Ferriscan) showed normal liver iron of 1.7 mg/g dry weight (reference range 0.17–1.8 mg/g).
Case 2
A man aged 75 years had originally presented with late-onset cataracts and persistent hyperferritinaemia (1200 μg/L). Serum iron and transferrin saturation were normal. His mother had had early-onset cataracts but not hyperferritinaemia. Initially, HHCS was suspected.
Percentage glycosylated ferritin in serum was determined for these two cases and four other patients (three from one family: two siblings and son of one sibling; and one individual patient), and all found to have unexplained hyperferritinaemia. A detailed history of early-childhood illnesses for these individuals had not been taken and it was only later realized that this second group of four patients had all had bilateral cataracts removed in early life. Two of these four had undergone liver biopsy during investigation of hyperferritinaemia.
Determination of percentage glycosylated ferritin
Glycosylated ferritin was determined using the method of Worwood et al. 8 with the following modification. Sepharose 4B-concanavalin A (Sigma-Aldrich, Gillingham, UK) was washed in 50 mmol/L sodium barbitone buffer in 500 mmol/L saline pH 8.0. To 0.2 mL serum, 1.5 mL of sepharose 4B-concanavalin A (sepharose suspension in buffer 1:1 by volume) was added followed by 0.3 mL barbitone buffer in stoppered tubes. The mixtures were incubated on roller mixers for two hours, and were then centrifuged at 3000 rpm for 10 min. Serum controls were run using sepharose 4B i (Sigma-Aldrich) in barbitone buffer in place of the sepharose/concanavalin A conjugate. Ferritin was measured in the supernatant using a Siemens Centaur ferritin immunoassay (Siemens Healthcare Diagnostics Ltd, Frimley, UK) (this manufacturer utilizes a sodium barbital buffer reaction matrix for ferritin assay: sample dilution in barbitone buffer gave satisfactory recoveries). Percentage glycosylation was calculated by comparing tests with controls. All analyses were carried out in duplicate.
DNA analysis
DNA sequencing was carried out as described by Kannengiesser et al. 6 All patients gave informed consent.
Results
Serum glycosylated ferritin was markedly elevated in both cases 1 and 2: 95% and 98%, respectively (reference range 50–81%). Hyperglycosylation of ferritin has been reported previously in association with hyperferritinaemia due to a mutation (c.89C>T) in the gene encoding the FTL in the heterozygous state, resulting in replacement of isoleucine by threonine at position 30 in the A helix (p.Thr30Ile).
6
It was anticipated that these two patients would carry the same mutation, but DNA sequencing revealed two different novel missense mutations. Case 1 is a carrier of an undocumented missense mutation in the heterozygous state, valine replacing alanine at position 27 of the A helix in the FTL (c.80C>T, p.Ala27Val). Case 2 is also heterozygous for a further undocumented missense mutation, leucine replacing glutamine at position 26 of the A helix in the FTL (c.77A>T, p.Gln26Leu). Neither amino acid variation was found in the Seattle SNPs database (
The p.Gln26Leu mutation was also found in another patient in a cohort of unexplained hyperferritinaemia cases referred to Bichat Hospital. Although glycosylated ferritin was not measured for this individual, the detection of this mutation further implicates L ferritin missense mutations in benign hyperferritinaemia.
In the remaining four patients, the percentage of serum glycosylated ferritin was low. Values of 20%, 28% and 42% for the family group of three were found with corresponding serum ferritin concentrations of 1822 , 1500 and 1718 μg/L, respectively. At this stage, the history of childhood cataracts became evident and confirmation of the HHCS was established with detection of a ‘classical’ mutation of the IRE of the FTL, a + 32G>C (c. − 168G>C) heterozygous mutation as previously described. 9 Serum ferritin was 37% glycosylated in the individual patient with serum ferritin of 1774 ug/L. This patient is heterozygous for another previously described mutation of the IRE of the FTL, a + 36C>A (c. − 164C>A). 10
Discussion
Hyperglycosylation of serum ferritin (>90%) strongly suggests the presence of a mutant FTL, as no other conditions have been reported consistently to lead to increased percentage glycosylation. This simple analysis appears to be an effective initial biochemical screen to identify patients for whom sequencing of the FTL is likely to be productive. In their previous study, Kannengiesser et al. 6 found perfect co-segregation between the mutated allele and hyperferritinaemia.
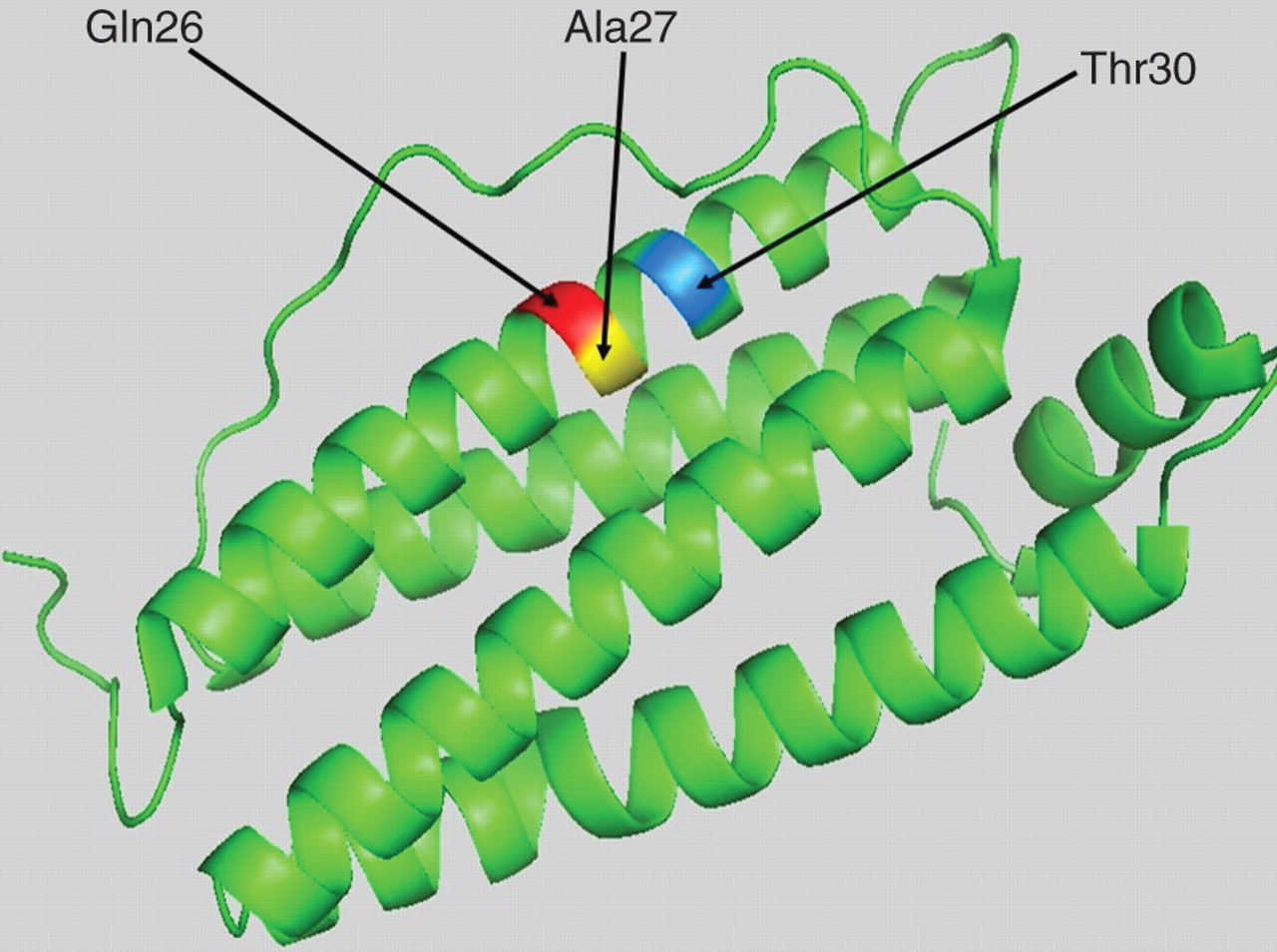
As shown with the previously published missense mutation of the FTL (p.Thr30Ile), the new mutations reported here at positions 26 and 27 of the A chain, extend the hydrophobic cluster (Figure 1) at the N terminal of the FTL. Since glutamine 26 and alanine 27 are in the same vicinity as the threonine 30 (Figure 2), hyperglycosylation may arise by a shared mechanism. The reason for development of hyperferritinaemia and hyperglycosylation associated with these mutant ferritin forms is not established. It is possibly related to increased secretion,
6
but delayed clearance may also contribute. Cragg et al.
11
demonstrated that I-131-labelled spleen ferritin (unglycosylated) was rapidly cleared from the circulation in two volunteers, whereas I-131-labelled plasma ferritin obtained from patients with haemochromatosis (>50% glycosylated) was cleared much more slowly (t
1/2 50 h compared with 5 hours).
Consequences of the missense mutations p.Gln26Leu and p.Ala27Val on the hydrophobicity of the N terminus of L ferritin. Hydrophobicity cluster analysis of the A helix in the wild-type L ferritin, suggests that the missense mutations p.Gln26Leu and p.Ala27Val extend the hydrophobic cluster (hydrophobic residues are indicated in grey and the hydrobic cluster is encircled). The amino acid code is that used with the International Nucleotide Sequence Database Position of Gln26, Ala27 and Thr30 in the L ferritin crystal. Amino acid residues are on the A helix. The crystallographic data of L ferritin subunit PDB 2FFX were analysed using Pymol software (Schrödinger Inc, Portland, NY, USA)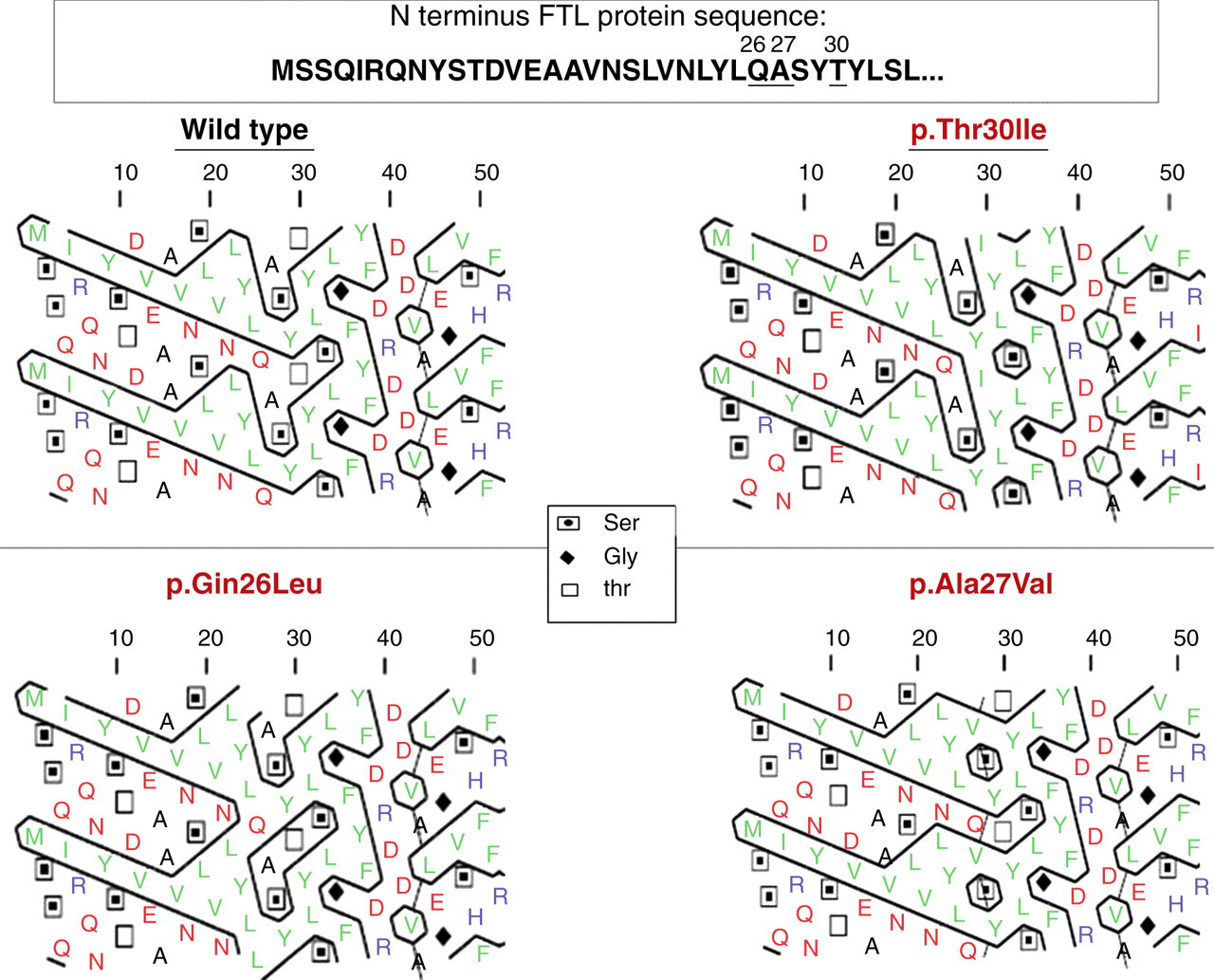
The incidence of variant ferritin forms is unknown. Kannengiesser et al. found an incidence of approximately 20% for the p.Thr30Ile variant among patients with unexplained hyperferritinaemia without iron overload. It seems likely, however, that other as yet unidentified mutations in the FTL will be found in cases of unexplained hyperferritinaemia.
Hyperglycosylation of the variant ferritin forms contrasts with the findings for the HHCS patients for whom percentage glycosylation was consistently low. There are a few published values for percentage glycosylation of serum ferritin in HHCS, but the low values obtained here (20–42%) are broadly consistent with those found in another study of seven affected individuals (30–55%). 12
Conclusion
It is important to consider genetic causes of hyperferritinaemia in healthy individuals presenting with persistently raised serum ferritin, but normal serum iron and transferrin saturation.
Measurement of percentage glycosylated ferritin is a simple procedure that may be used to unmask genetic forms of hyperferritinaemia. In particular, hyperglycosylation (>90%) suggests a benign form of hyperferritinaemia that requires no treatment and no further investigation.
DECLARATIONS