
Abstract
Ammonia is produced continuously in the body. It crosses the blood–brain barrier readily and at increased concentration it is toxic to the brain. A highly integrated system protects against this: ammonia produced during metabolism is detoxified temporarily by incorporation into the non-toxic amino acid glutamine. This is transported safely in the circulation to the small intestine, where ammonia is released, carried directly to the liver in the portal blood, converted to non-toxic urea and finally excreted in urine. As a result, plasma concentrations of ammonia in the systemic circulation are normally very low (<40 μmol/L). Hyperammonaemia develops if the urea cycle cannot control the ammonia load. This occurs when the load is excessive, portal blood from the intestines bypasses the liver and/or the urea cycle functions poorly. By far, the commonest cause is liver damage. This review focuses on other causes in adults. Because they are much less common, the diagnosis may be missed or delayed, with disastrous consequences. There is effective treatment for most of them, but it must be instituted promptly to avoid fatality or long-term neurological damage. Of particular concern are unsuspected inherited defects of the urea cycle and fatty acid oxidation presenting with catastrophic illness in previously normal individuals. Early identification of the problem is the challenge.
Introduction
Ammonia is produced continuously in the body. It crosses the blood–brain barrier readily and at increased concentration it is toxic to the brain. A highly integrated system protects against this: ammonia produced during metabolism is detoxified temporarily by incorporation into the non-toxic amino acid glutamine. This is transported safely in the circulation to the small intestine, where ammonia is released, carried directly to the liver in the portal blood, converted to non-toxic urea and finally excreted in urine. As a result, plasma concentrations of ammonia in the systemic circulation are normally very low (<40 μmol/L). Hyperammonaemia develops if the urea cycle cannot control the ammonia load. This occurs when the load is excessive, portal blood from the intestines bypasses the liver and/or the urea cycle functions poorly. By far, the commonest cause at all ages is liver damage associated with abnormal liver function, often with portosystemic shunting. This review focuses on other causes in adults. Because they are much less common, diagnosis may be missed or delayed, with disastrous consequences. There is effective treatment for most of them, but it must be instituted promptly to avoid fatality or long-term neurological damage. Of particular concern is the inherited urea cycle defect (UCD), ornithine transcarbamylase deficiency. There is an escalating number of reports of fatal hyperammonaemic crises in previously asymptomatic adults who could have made a good recovery had the diagnosis been made at presentation.
Ammonia: sources and detoxification by glutamine and the urea cycle
Ammonia
Ammonia is generated during metabolism in all organs. Large amounts are produced in skeletal muscle, particularly during exercise, mainly from deamidation of adenosine monophosphate, with some from amino acid catabolism. These processes and glutamate dehydrogenase activity contribute to ammonia production in the brain. 1,2 Another important source is release from the kidneys during the production of ammonia to buffer H+ ions in urine. Glutamine is taken up from the blood and metabolized by phosphate-dependent glutaminase in the proximal tubular cells, releasing ammonia. Normally, approximately 30% of total renal ammonia is excreted with H+ ions. The other 70% is released into the circulation. This situation changes dramatically with acute disturbances of acid–base balance: the percentage of ammonia excreted in urine can range from 50% to 70% in acute acidosis to 18% in alkalosis. 3,4 Ammonia is also produced by bacterial degradation of urea and amino acids in the lumen of the large bowel. This accounts for around 50% of ammonia produced in the bowel when fasting, but more after protein feeding or gastrointestinal bleeding. 3,4
Glutamine
Glutamine is a non-toxic/non-essential amino acid which constitutes approximately 50% of the whole-body free amino acid pool and normally is the most abundant amino acid in the plasma and cerebrospinal fluid (CSF). Through metabolism to 2-oxoglutarate, a tricarboxylic acid cycle intermediate, it is an obligatory fuel for rapidly dividing cells, notably of enterocytes in the small intestine and cells of the immune system. In addition to its role in renal regulation of acid–base balance, it is central to the processes involved in ammonia detoxification. This is achieved by the concerted actions of two enzymes. Glutamine synthetase (GS) combines glutamic acid with ammonia to form glutamine in an adenosine 5′-triphosphate (ATP)-consuming reaction. Phosphate-activated glutaminase (PAG), in contrast, degrades glutamine to glutamic acid and ammonia.
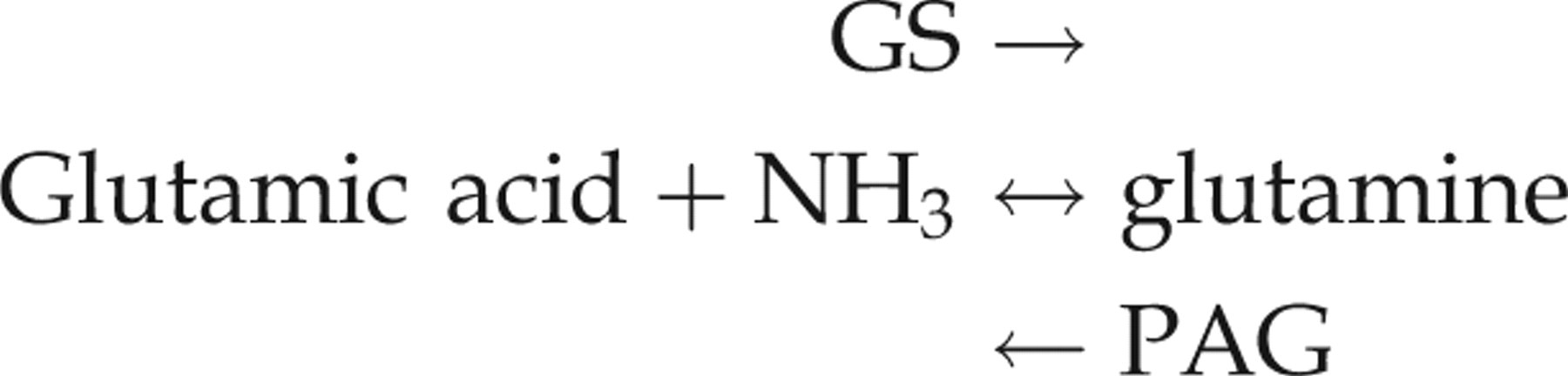
According to the relative activities of these enzymes, glutamine either removes ammonia (high GS) or is a source (high PAG). The mucosa of the small intestine produces a large amount of PAG and little GS, and hence has a high capacity to generate ammonia. In liver, PAG is present in periportal hepatocytes which also contain the urea cycle. Cells around the hepatic venules at the centre of liver acini, on the other hand, contain GS. They can scavenge and detoxify ammonia which has escaped the urea cycle, preventing its release into the systemic circulation. In brain, there is also compartmentalization of the enzymes. Astrocytes contain around 80% of brain GS and can rapidly incorporate ammonia into glutamine. This is then transferred to neurons. Within neurons, PAG degrades glutamine to glutamate which is secreted into neural synapses to function as a neurotransmitter. Astrocytes re-claim secreted glutamate from the synaptic clefts and re-synthesize glutamine. Muscle expresses only small amounts of GS, but because of its large mass, has significant capacity to detoxify ammonia. 3–6
Coordination of glutamine metabolism to detoxify ammonia
Glutamine generated in the tissues is released into the circulation. Some is extracted by the kidneys and cells of the immune system. Most, however, is taken up by the small intestinal cells and metabolized by PAG to ammonia and glutamate which is oxidized for energy or metabolized to citrulline. Citrulline and ammonia are carried in the portal blood stream to the liver, along with ammonia produced by bacterial activity in the colonic lumen (Figure 1). Ammonia concentrations in portal blood are 5–20 times higher than in the systemic circulation. Most of the ammonia is converted to urea in the periportal hepatocytes. Residual ammonia is detoxified by GS in perivenous hepatocytes. Citrulline passes through the liver into the blood, is taken up by the kidneys and incorporated into arginine.
7–9
Role of glutamine in the short-term buffering of ammonia produced in the body. NH3, ammonia; Glmn, glutamine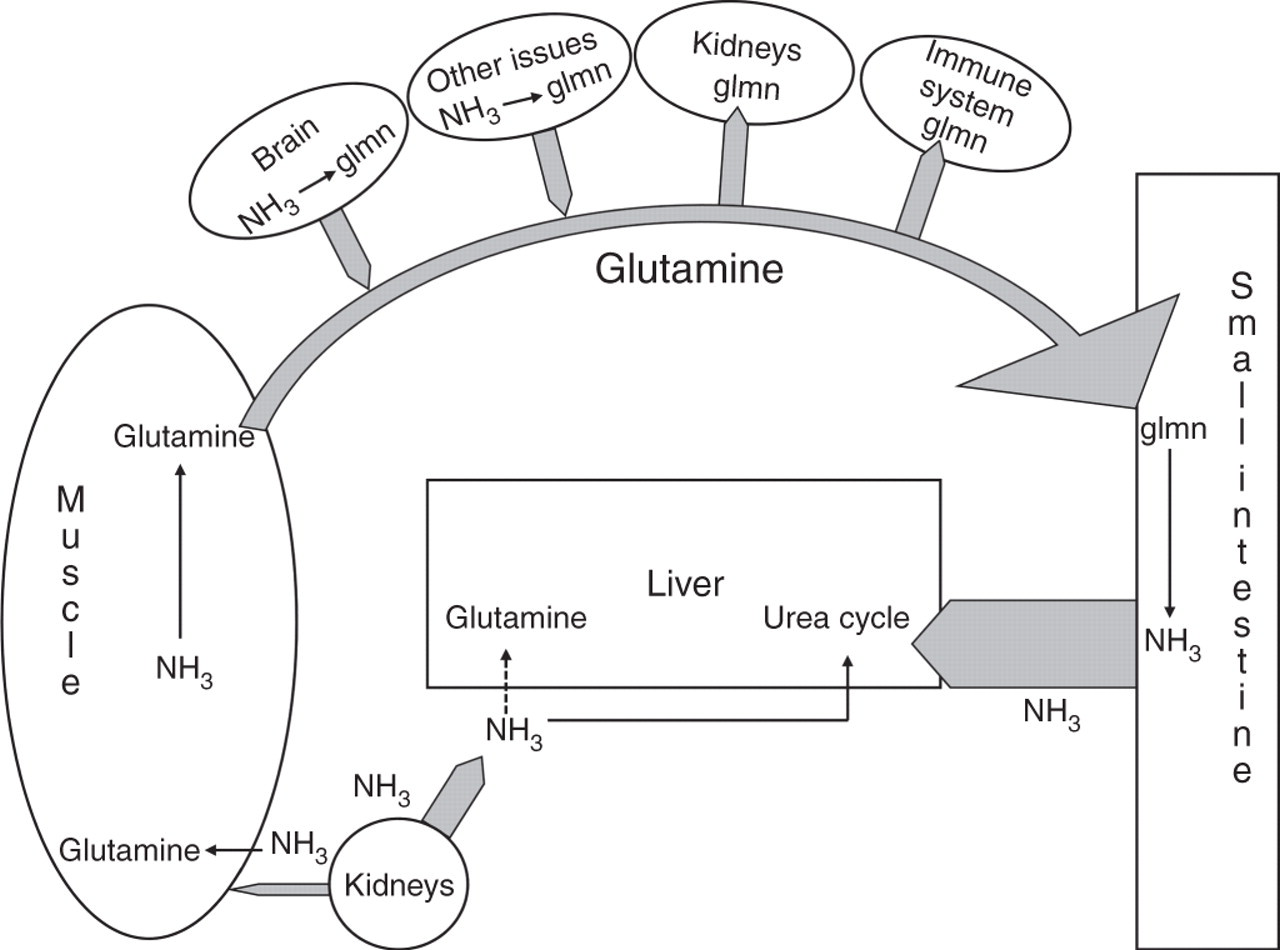
The urea cycle
In humans, the urea cycle is the major route through which ammonia and surplus amino-group nitrogen are removed from the body. It also produces arginine by de novo synthesis. The cycle comprises six enzyme reactions (Figure 2): the first three are intramitochondrial and the others are cytosolic. Carbamyl phosphate synthetase I (CPS I) has an absolute requirement for N-acetylglutamate (NAG), an allosteric activator produced from acetyl-CoA and glutamate by NAG synthetase. The urea cycle substrates are ammonia, bicarbonate and aspartate. Ornithine acts as a carrier and is not degraded. An adequate supply of ornithine and arginine is critical for the cycle to function normally. With each turn of the cycle, two atoms of nitrogen (N) are converted to urea, one from ammonia and another from the amino acid pool, channeled through aspartate. Alanine is a major source. Its amino group is first transferred by transamination to 2-oxoglutarate, forming glutamate, then on to oxaloacetate producing aspartate. The liver is the only organ with the full complement of urea cycle enzymes. Normally, the cycle operates at around 20–50% of maximal capacity. Short-term regulation is through the action of NAG on CPS I and reflects the availability of acetyl-CoA, glutamate and arginine, the activator of NAG synthetase. Long-term control is achieved through altering enzyme production, possibly by transcription factors and hormonal regulation.
2,10–13
The urea cycle. NAG, N-acetylglutamate; NAGS, N-acetylglutamate synthetase; CPS I, carbamyl phosphate synthetase I; OTC, ornithine transcarbamylase; AS, argininosuccinic acid synthetase; AL, argininosuccinic acid lyase; α-KG, 2-oxoglutaric acid; 1, mitochondrial ornithine transporter (ORNT I); 2, aspartate-glutamate carrier (citrin); 3, 2-oxoglutarate transporter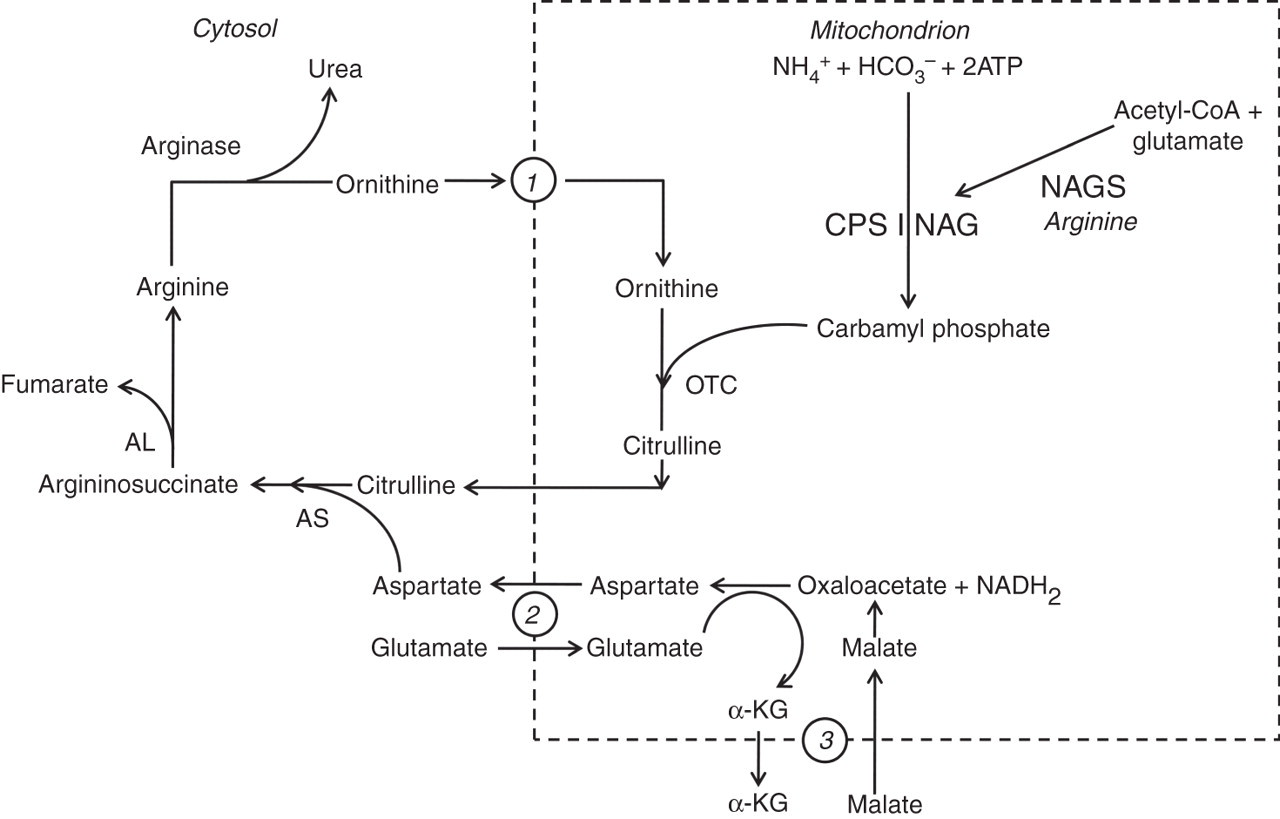
Orotic acid
Orotic acid is an intermediate in pyrimidine biosynthesis in the cytosol. It is normally produced from carbamyl phosphate synthesized by cytosolic CPS II from glutamine and bicarbonate using ATP. However, when mitochondrial carbamyl phosphate accumulates because it is not used in the urea cycle, it diffuses into the cytoplasm and is pulled into the pyrimidine pathway, increasing the production of orotic acid and other pyrimidines (Figure 3). This situation arises when high concentrations of arginine increase NAG synthesis and hence the activity of CPS I, or when there is a defect in the urea cycle after carbamyl phosphate preventing its use. This could result from an inherited defect of the urea cycle or of ornithine transport or to a deficiency of arginine, the main ornithine precursor (Table 1). Normally very little orotic acid is excreted in urine.
14,15
Diversion of mitochondrial carbamyl phosphate to pyrimidine synthesis in the cytosol. NAG, N-acetylglutamate; CPS I, carbamyl phosphate synthetase 1; CPS II, carbamyl phosphate synthetase II; OTC, ornithine transcarbamylase; •, orotidine-5′-phosphate (OMP) decarboxylase, the enzyme inhibited by the allopurinol metabolite oxypurinol ribonucleotide Causes of orotic aciduria OTC, ornithine transcarbamylase; AS, argininosuccinic acid synthetase; AL, argininosuccinic acid lyase; NAGS, N-acetylglutamate synthetase; HHH, hyperornithinaemia, hyperammonaemia, homocitrullinuria syndrome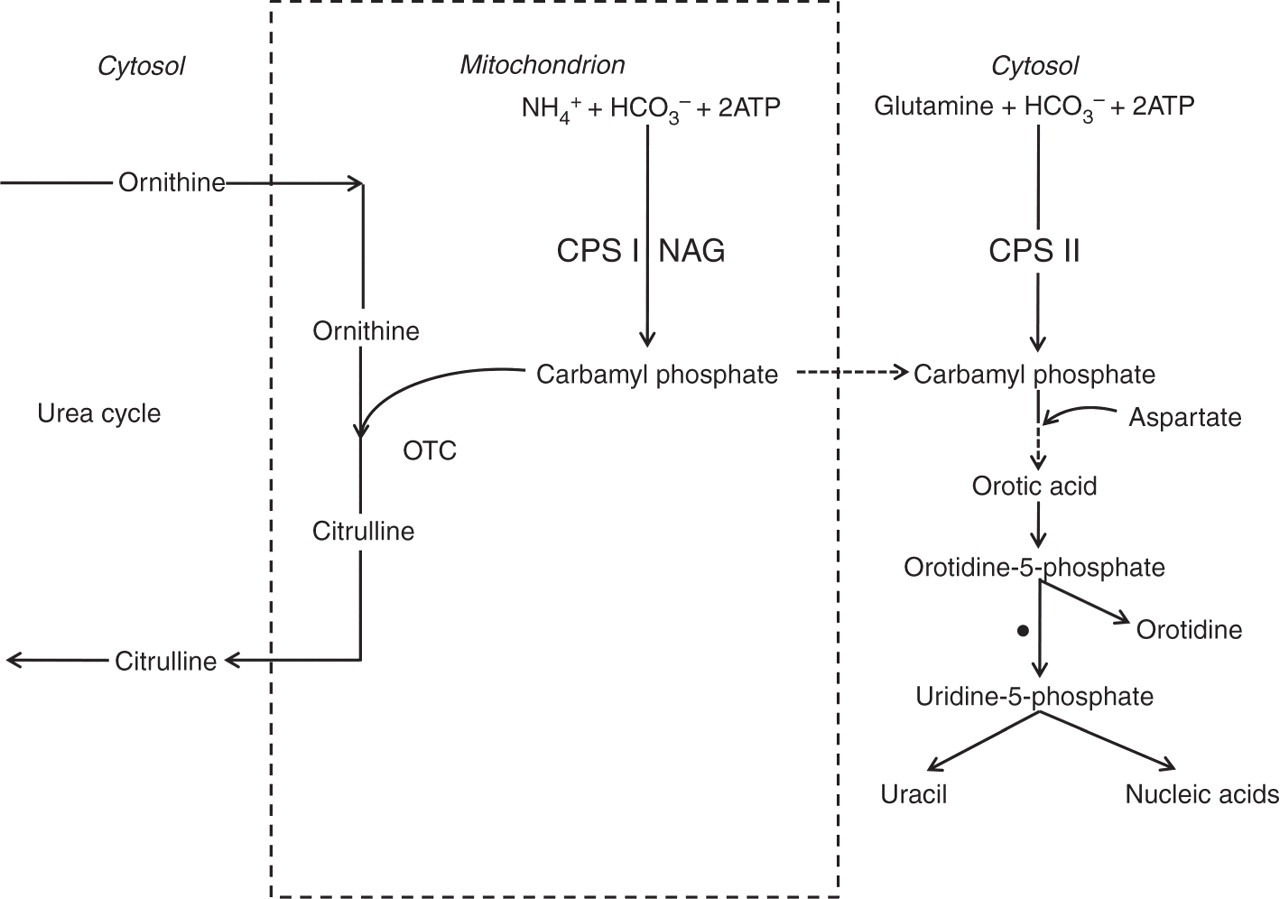
Toxicity of ammonia
In body fluids, gaseous ammonia (NH3) is in equilibrium with ionic ammonium (NH4
+).
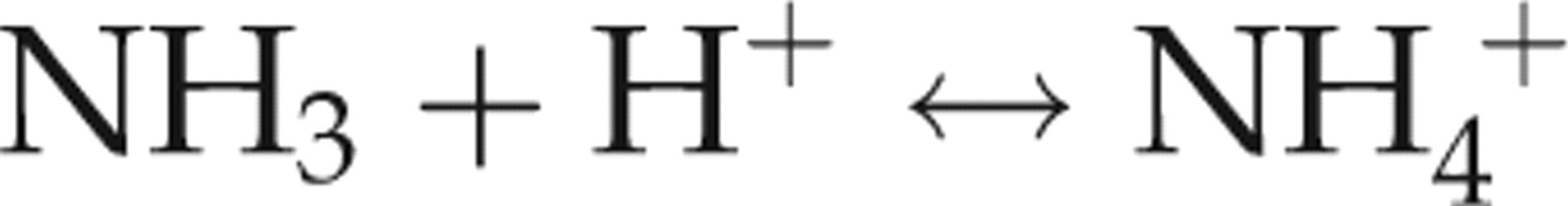
At 37°C, the pKa of NH3 is 9.15, and hence at the normal physiological pH of 7.4, 98% is present as NH4 +. The proportion of NH3 is higher in systemic alkalosis. Cell membranes are five times more permeable to NH3 than NH4 +. Since most laboratories analyse total ammonia (NH3 + NH4 +), ‘ammonia’ in the clinical sections of this review includes both. Normally, ammonia concentrations in brain are 1.5–3.0 times higher than in blood. Some is produced by brain metabolism and some derives from the circulation. NH3 is lipid soluble and enters the brain by diffusion. NH4 + enters through membrane ion channels and transporters. It has similar ionic properties to K+ and can compete with K+ for transport. In addition, it may enter through the aquaporin 8 channel and there is evidence for a specific ammonia transporter. 1,2,16
Initial adaptation to hyperammonaemia
When ammonia is not cleared by the urea cycle, increased glutamine production in other organs temporarily protects against toxicity. In experimental hyperammonaemia, skeletal muscle uptake is correlated with arterial ammonia concentrations and muscle is a major site for ammonia removal. The increased ammonia entering the brain from the circulation is metabolized almost exclusively to glutamine. However, this protection is limited since astrocyte GS functions at near maximal capacity under normal physiological conditions. The kidneys respond to hyperammonaemia initially by excreting more ammonia in urine and absorbing less into venous blood. This adaptation fails as hyperammonaemia intensifies, and the renal contribution to circulating ammonia increases. 2–5,17
Clinical features of toxicity
Clinical features of ammonia toxicity
Mechanisms of toxicity
Abnormalities which may contribute to ammonia toxicity
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA, N-methyl-
Accumulation of glutamine in astrocytes is probably implicated in acute cerebral oedema. This is usually attributed to its osmotic action. In chronic or intermittent hyperammonaemia, astrocytes expel myo-inositol and other weaker osmolytes to maintain osmotic equilibrium. This compensation is ineffective in rapidly developing severe hyperammonaemia. Glutamine has also been shown to increase the permeability of the inner mitochondrial membrane and to initiate free radical damage. These actions may contribute to oedema. The significance of other mechanisms proposed to explain cerebral oedema is uncertain. 12,22,25,26
A significant early event in hyperammonamia could be accumulation of extracellular potassium due to competition of NH4 + ions with K+ on K+-co-transporters and exchangers and down-regulation of potassium channels, observed in acute hyperammonaemia. An increase in resting membrane potential with depolarization of neurons might increase glutamate excitotoxicity. 16,24,27
Ammonia has a number of actions which could disturb glucose metabolism in brain (Table 3) and collectively increase brain lactate and compromise energy production. However, these may be secondary contributory disturbances since changes in ATP and creatine phosphate were not detected using 32P nuclear magnetic resonance in acute or chronic hyperammonaemia, except preterminally. 1,6,10,11
There are numerous disturbances in neurotransmitter systems, including increased turnover of 5-hydroxytryptamine. 28 These are undoubtedly central to the pathogenicity of ammonia, but are likely to be cascaded secondary sequelae.
Low concentrations of glutamic acid and glutamine may explain why persistent increases in plasma ammonia do not cause neurotoxicity in infants with hyperinsulinaemic hyperammonaemia due to a genetic increase in glutamate dehydrogenase. 29
Causes of hyperammonaemia not due to liver disease
Causes of non-hepatic hyperammonaemia in adults
*With other predisposing factors
Inherited UCDs
Diagnostic abnormalities of inherited urea cycle and transporter defects
*Glutamine and alanine are commonly increased and are useful indicators of a urea cycle defect but are non-specific
†Argininosuccinic acid is not found in normal plasma and urine
OTC deficiency
OTC deficiency accounts for the majority of adult cases of UCD presenting with acute encephalopathy and is the commonest UCD. The OTC gene is located on the X-chromosome at Xp21 and is expressed in the liver and gut. Around 350 pathological mutations have been identified, and currently a mutation is identifiable in about 80% of cases. 34 The clinical phenotype in affected men and female carriers shows a spectrum of severity, even within the same family. Usually, boys are severely affected and present neonatally, but some have a mild disorder which may not be obvious clinically. The severity in girls depends on the mutation and on the proportion of hepatocytes expressing the mutant allele. Only 15% of female carriers have symptoms. 30 Girls with unfavourable expression may present as neonates. Asymptomatic carriers have normal IQ scores, 35,36 but recent magnetic resonance spectroscopy studies have found white matter changes in tracts involved in memory and executive function. 25 They may still have hyperammonaemic crises. In one study, the first occurred in 19% of women after 16 y of age. 23
With growing awareness, UCDs are being recognized increasingly in previously apparently well adults presenting with acute encephalopathy, often fatal if treatment is delayed. Although most have OTC deficiency, NAGS, CPS I and AS deficiencies have also been reported. Hyperammonaemic crises have been precipitated by increased protein intake, glycine irrigation of the bladder, infections, surgery, chemotherapy, parenteral nutrition, gastrointestinal bleeding and sodium valproate. 2,37–47 Diagnosis may be delayed if there are co-morbid conditions such as trauma. 43 The postpartum period (3–8 d) is a particularly dangerous time because of collagen and smooth muscle breakdown as the uterus involutes. 48 Previously asymptomatic and undiagnosed women have developed severe hyperammonaemic encephalopathy following a normal pregnancy. 12,49–51
Hyperammonaemia, hyperornithinaemia, homocitrullinuria syndrome
Hyperammonaemia, hyperornithinaemia, homocitrullinuria (HHH) syndrome is due to mutation of the SLC25A15 gene which encodes for ornithine translocase, which transports ornithine into mitochondria. The low intramitochondrial ornithine decreases urea cycle activity, causing hyperammonaemia, and carbamyl phosphate accumulates. Some combines with lysine instead of ornithine, producing homocitrulline which is excreted in urine, and some is incorporated into orotic acid. Patients have protein intolerance. Most present in childhood with chronic neurological problems, sometimes with acute liver dysfunction and coagulopathy. Adults have presented with hyperammonaemia. 52,53
Lysinuric protein intolerance
Lysinuric protein intolerance is caused by mutations in the SLC7A7 gene which codes for the carrier for the basic amino acids, ornithine, arginine and lysine and is expressed in epithelial cells of the kidneys and intestine. Plasma ammonia and urinary orotic acid may be normal when fasting, but increased for several hours after a protein meal. In addition to protein intolerance, patients may have osteoporosis, liver disturbance and lung, renal and autoimmune disease. Adults have presented in hyperammonaemic coma. 54–57
Citrin deficiency (citrullinaemia type II)
Citrin deficiency (citrullinaemia type II) is due to mutation of SLC25A13 which encodes for citrin, an aspartate–glutamate carrier expressed in the inner mitochondrial membrane of hepatocytes. It transports glutamate into mitochondria and aspartate out. It is a component of the malate–aspartate shuttle which carries reducing equivalents into mitochondria, and through this has roles in producing and degrading simple sugars and nucleotides and in protein synthesis. It also supplies cytosolic aspartate for the urea cycle. In the adult form (citrullinaemia type II), patients present at around 20–50 y of age with recurring episodes of hyperammonaemia which may be fatal. There is fatty infiltration of the liver but little or no liver dysfunction. Most are thin, with an aversion to high carbohydrate foods and select a protein- or lipid-rich diet. Most cases have been among the Japanese, but the disorder occurs worldwide. 58
Diagnosis of UCDs
The key diagnostic tests to differentiate the urea cycle and transport defects are plasma and urine amino acids and urine orotic acid (Table 5). Plasma glutamine and alanine are often raised non-specifically and are useful alerting clues. Diagnosis is then confirmed by enzyme analysis of fibroblasts or liver and/or mutation analysis. Liver enzyme analyses are often problematic and biopsy is risky. It may be avoided if metabolite abnormalities are clear-cut or, if not, by molecular diagnosis if available. Detection of OTC deficiency in asymptomatic carriers is difficult. Although fasting glutamine and ammonium and alanine concentrations of asymptomatic carriers are significantly higher and citrulline and arginine lower than those of normal control women, they are often within normal limits and not diagnostic. 35 If the mutation is known, DNA testing is recommended. Protein load and alanine loading tests are possibly risky and are unreliable, with too many false-positives and -negatives. Measurement of ureagenesis by stable isotope infusion is better, but this test is not widely available. The allopurinol loading test is safe and can be performed at home. In this, a metabolite of allopurinol, oxypurinol ribonucletide, blocks the pyrimidine biosynthetic pathway leading to the accumulation of orotidine and orotic acid. Urinary concentrations of these metabolites are increased in OTC carriers. Although an early study reported a sensitivity of 95.8% and a specificity of 88%, recent data indicate that the test is less reliable. 59–61
Management of UCDs
In acute hyperammonaemia, treatment should be started before the precise diagnosis is made. Advice from a tertiary centre must be sought promptly. Published detailed guidelines are available and in the UK are accessible on the website of the British Inherited Metabolic Disease Group.
11,12,33,62–65
The aims are to reduce production of nitrogenous waste and to lower plasma ammonia concentrations before cerebral oedema develops. This is achieved by withdrawing all protein for 24–48 h, supplying a hypercaloric, protein-free solution given with insulin to enhance anabolism, an infusion of arginine, exploiting alternative pathways for excretion of waste nitrogen (see below), and dialysis.
Haemodialysis reduces plasma ammonia more quickly than haemofiltration or peritoneal dialysis. Exchange transfusion only clears toxins from the circulation and is potentially dangerous. Dialysis removes nutrients from plasma, creating a catabolic state, and plasma ammonia may rebound unless coupled with aggressive nutritional management. 66
The mainstays of long-term management are dietary protein restriction, arginine or citrulline supplements, and oral alternative pathway medications to augment nitrogen excretion. The diet must supply enough protein-free energy to prevent net catabolism of body proteins and should be supplemented with minerals, vitamins and trace elements. Except in arginase deficiency, arginine supplementation is important because synthesis by the urea cycle is blocked and it becomes an essential or semi-essential amino acid. It is needed for creatine and nitric oxide synthesis. Citrulline may be substituted in severe CPS I and OTC deficiencies and triple H syndrome. In severe defects, a mixture of essential amino acids higher in branched chain amino acids may be incorporated. Fasting plasma concentrations of ammonium, branched chain amino acids, arginine and protein should be maintained within normal limits and glutamine <1000 μmol/L, ideally <800 μmol/L. Plasma glutamine seems to predict hyperammonaemia, and is a guide to the effectiveness of treatment. An emergency regimen supplying a high calorie/low protein intake is instituted when at risk. Treatment must be lifelong unless liver transplantation is undertaken. Elective surgery, anaesthesia and pregnancy require careful management to avoid decompensation. 12,33,62,64,66 In contrast to UCDs, the diet for citrin deficiency is high in protein and lipid and low in carbohydrate. 58
Alternative pathway therapies
These promote the synthesis of nitrogen-containing metabolites with high urinary excretion rates as alternatives to urea for removing waste nitrogen from the body. This leaves the urea cycle with spare reserve capacity and permits an increase in dietary protein intake. Three agents are used currently: sodium benzoate, sodium phenylacetate (usually as its precursor, sodium phenylbutyrate) and arginine in high doses (AS and AL deficiencies). 2,11,67
Sodium benzoate is conjugated rapidly with glycine to form hippurate which is excreted. More glycine is re-synthesized from serine which, in turn, is replaced using nitrogen derived from alanine or glutamine. In theory, each mole of benzoate administered could remove one mole of waste nitrogen. However, this is not achieved.
Phenylacetate is conjugated with glutamine to form phenylacetylglutamine (PhGmn), in the liver and kidneys, and, excreted. Glutamine is continuously replenished by amidation of glutamic acid with ammonia by GS. Since glutamine contains two nitrogen atoms, one mole of phenylacetate could potentially remove two moles of nitrogen. However, the recovery of PhGmn is variable, ranging from 50% to 90% of administered dose. Phenylacetate has an objectionable clinging odour and is now generally given as its odourless precursor, sodium phenylbutyrate.
In addition to its use as a replacement substitute in UCDs, arginine is given in large doses as an alternative pathway treatment in AS and AL. It is converted to ornithine in the urea cycle and provides both enough ornithine to top up the urea cycle, and surplus ornithine to combine with aspartate and remove waste nitrogen as citrulline in AS deficiency and arginininosuccinate in AL deficiency. Both are readily excreted in urine.
Sodium phenylbutyrate may cause painful mucositis of the oesophagus and stomach. Otherwise, there are no serious side-effects when the drugs are used in the correct doses. Excessive doses of sodium benzoate and sodium phenylacetate are toxic.
68
The drugs should be used cautiously if liver function is impaired since phenylacetate and benzoate conjugation occur in liver. Renal function should be monitored since efficacy depends on renal clearance of the metabolites. Carnitine combines with benzoate and inactivates it,
69
and penicillins and other antibiotics may compete with PhGmn and hippurate for excretion. If locally available, monitoring plasma benzoate and phenylacetate concentrations may help to reduce toxicity. The safety of phenylbutyrate in pregnancy is unknown and until this is established, use of sodium benzoate, which is a normal dietary constituent, may be preferable.
31
Trials of a novel scavenging agent, glyceryl tri (4-phenylbutyrate) are in progress,
70
and early studies of
Other treatments
N-carbamyl-
All families with a diagnosed UCD should receive genetic counselling. Increasingly, diagnosis and carrier status can be ascertained by DNA analysis. All six UCDs can be diagnosed antenatally by chorionic villus biopsies taken at 9–11 weeks of gestation. Successful preimplantation genetic diagnosis for OTC deficiency has been reported. 77
Fatty acid oxidation defects
In these disorders, energy is not produced efficiently from long-chain (C16–C22) fatty acids because of a defect in the process of mitochondrial β-fatty acid oxidation: a defect in the carnitine cycle may interfere with fatty acid import into mitochondria, or one of the β-oxidation enzymes may be deficient. Fatty acid oxidation is an essential source of energy for the liver, for contraction of heart and for skeletal muscle and, during fasting, supplies ketones as an alternative fuel to glucose for the brain, kidneys and muscles. The components of the carnitine cycle are the
Inherited defects occur at all steps of fatty acid oxidation. Depending on the site, these present with metabolic crises after fasting with acute encephalopathy and liver disturbance, cardiomyopathy and rhabdomyolysis, in variable combinations. Hyperammonaemia is a feature of metabolic crises. These occur with deficiencies of the carnitine transporter, CPT I, translocase, CPT II, VLCAD, LCHAD, trifunctional protein and MCAD, generally in infants or young children who have fed poorly for 24–36 h. Lipolysis and fatty acid oxidation are activated but ketone production is severely impaired. Typically, lethargy, nausea and vomiting progress rapidly to coma and brain stem failure within one or two hours. The liver may enlarge. Hypoglycaemia is a late feature and its absence does not exclude a fat oxidation defect and neither does ketonuria, which is often present acutely. Hyperammonaemia has been attributed to decreased synthesis of NAG because of a poor supply of acetyl-CoA from fatty acid oxidation, as well as to liver disturbance. Mortality is high and survivors may be brain-damaged, but the outcome may be excellent with early intervention. In populations of European descent, the commonest defect is MCAD deficiency (MCADD) and there is a common allele (C985A>G), regarded as a severe mutation. In the UK, the prevalence of MCADD from neonatal screening of 1.4 in 10,000 is higher than the incidence of around one in 10,000 from those presenting clinically. Hence, a significant number of unscreened individuals remain undetected. 78–82
Tolerance to fasting increases with age. However, previously healthy adults with an unsuspected fatty acid oxidation defect have presented with an acute hypoketotic encephalopathy and sometimes rhabdomyolysis after an unusual fasting stress, in some associated with alcohol or pregnancy. The majority have had MCADD, with a mortality of around 50%, which is higher than in children at first presentation (23%). VLCAD deficiency and multiple acyl-CoA dehydrogenase deficiency have also been reported. Metabolic decompensation during pregnancy also occurs in women carrying a fetus who is homozygous for LCHAD deficiency. It is probable that cases are missed. 80,83–86
The key diagnostic tests are blood spot or plasma acylcarnitines and, at presentation, plasma free fatty acid:3-hydroxybutyrate ratio (high), plasma ammonia and urine organic acids. Confirmation is by enzyme analysis of skin fibroblasts and/or mutation analysis. 81,82,87 To avoid repeat crises, the priorities in MCADD are to avoid fasting, alcohol excess and dieting with rapid weight loss, and for close supervision of pregnancies and perioperative management. For other defects, low-fat diets supplemented with medium chain triglycerides and carnitine are used, with riboflavin in multiple acyl-CoA dehydrogenase deficiency.
Organic acid disorders
Severe hyperammonaemia complicates acute metabolic crises in inherited organic acid disorders of branched-chain amino acid catabolism. Urinary orotic acid is not increased. In most cases, the probable explanation is deficiency of NAG causing decreased activity of the urea cycle enzyme CPS I.
88–90
Abnormal acyl-CoA intermediates accumulate rapidly in these disorders, trapping coenzyme A. Initially, carnitine protects against this:
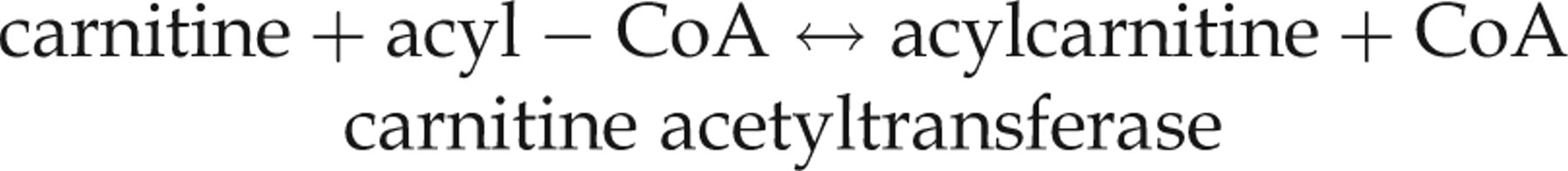
Propionyl-CoA inhibits NAG synthetase directly. This is likely to be important in propionyl carboxylase deficiency (propionic acidaemia), methylmalonic aciduria and multiple carboxylase and biotinidase deficiencies, in which propionyl-CoA accumulates. In addition, in these and the other branched-chain disorders (isovaleric acidaemia, β-ketothiolase and HMG-CoA lyase deficiencies), lack of acetyl-CoA for NAG synthesis is probably a major factor. This would arise because trapping of coenzyme A limits acetyl-CoA synthesis and carnitine depletion reduces its production by fatty acid oxidation. 10,88,90 Hyperammonaemia also occurs rarely in primary lactic acidosis. In severe deficiencies of pyruvate dehydrogenase, this is probably due to depletion of intramitochondrial acetyl-CoA. 91 In pyruvate carboxylase deficiency, hyperammonaemia is explained by decreased carboxylation of pyruvate to oxaloacetate which, after transamination to aspartate, is a substrate for the urea cycle. 92
Severe organic acid disorders usually present in infancy and have a poor outcome without rapid, appropriate intervention. However, young adults with less severe defects under good control do well and lead normal lives, although they are still at risk of decompensation during infections or other catabolic stress including surgery. 93 In addition, previously asymptomatic adults with isovaleric acidaemia have presented with acute crises. 94,95 Profound biotinidase deficiency has been detected in asymptomatic adults who may be at risk for acute decompensation. 96,97 Despite their rarity, organic acid disorders should be excluded in adults with acute encephalopathy without obvious cause.
The key diagnostic tests are analyses of plasma and urine amino acids, urine organic acids and blood acylcarnitines. Confirmation is by enzyme analysis of leukocytes or skin fibroblasts and/or mutation analysis. The synthetic analogue of NAG, N-carbamylglutamate, increased urea synthesis from [1-13C] acetate in hyperammonaemic patients with decompensated propionic acidaemia 98 and has reduced plasma ammonia in a small number of other babies with propionic acidaemia, and one with isovaleric acidaemia. 73 Use of drugs to increase waste nitrogen excretion are contraindicated because of the risk of toxic drug accumulation due to coenzyme A depletion. 63
Sodium valproate
Sodium valproate is used commonly to treat epilepsy and bipolar and affective disorders. Small transient increases in plasma ammonia to less than 2–3 times the upper limit of normal occur frequently with normal liver function, are usually asymptomatic and have no known consequences. This is more likely during combined therapy with other anticonvulsants. Transient increases occur after intravenous loading and are unrelated to plasma valproate concentration. Currently, there are no specific recommendations for screening for asymptomatic hyperammonaemia. 99,100
Valproate-induced hyperammonaemic encephalopathy is a rare and sometimes fatal complication. 100–103 It may occur acutely or during chronic therapy. Usually, liver function tests are normal and plasma valproate is within the therapeutic range and not correlated with daily dose or plasma ammonia. Plasma carnitine may be decreased, and urine orotic acid is normal, except in rare patients with an unsuspected UCD. Brain magnetic resonance spectroscopy shows markedly depleted myoinositol and choline. Hyperammonaemia is probably due largely to depletion of NAG due to sequestration of coenzyme A and to secondary carnitine deficiency, as occurs in inherited organic organic acidurias. The risk is increased by polytherapy with phenobarbital, phenytoin, carbamazepine, topiramate, pivmecillinam and salicylates (but not other non-steroidal anti-inflammatory drugs [NSAIDs]), carnitine deficiency and a UCD. Plasma ammonia must be checked if a progressive confusional syndrome or increased seizures occur after starting valproate. If raised, liver function tests and plasma carnitine should be analysed. Urine orotic acid and plasma and urine amino acids should be measured to investigate for a UCD. In addition to discontinuing valproate and supportive management, carnitine and N-carbamylglutamate may accelerate recovery. 103,104
Urinary tract disorders causing hyperammonaemia
Rarely, hyperammonaemic coma has developed when an anatomical or neurological abnormality of the lower urinary tract causes urinary stasis which predisposes to infection. If the bacterium produces urease (for example Proteus species, Corynebacterium species or Staphylococcus species), ammonia is produced from urea and the urine becomes alkaline (pH 8–9). Because the pKa of NH3 is 9.15, the ratio of NH3 to NH4 + increases to around 1:1, and free ammonia diffuses into the blood. 105,106
This situation may occur as a rare complication of surgical implantation of the ureters into the bowel in patients with congenital urological defects, neurological disturbances of bladder function or bladder or pelvic malignancies. In ureterosigmoidostomy, the ureters are implanted into the sigmoid colon and urine is excreted with the faeces. In ileal loop diversions, the ureters are implanted into a segment of ileum which has been surgically isolated from the rest of the bowel and opens onto the abdominal wall. Urine drains into a bag and is not mixed with faeces. Following ureterosigmoidostomy, ammonia formed from nitrogenous urinary constituents by colonic bacterial is absorbed into the portal blood. It is detoxified readily, providing the liver is functioning normally with a normal urea cycle, is not bypassed by portosystemic shunting or overwhelmed by an excessive ammonia load. This might occur with abnormal bacterial colonization of the colon, and/or constipation or bowel obstruction causing prolonged bowel transit time. Muscle wasting increases the risk of hyperammonaemia through decreased peripheral detoxification by GS. Hyperammonaemia with ileal loop diversions results from contamination of the urine with urease-producing bacteria. It appears to be extremely rare. One reported patient had an abnormally long ileal loop with recurrent urine infections. Ureterosigmoidostomy is now an uncommon operation, but there is an ongoing risk of hyperammonaemia from procedures to augment the bladder with bowel, which provide a large surface area of bowel mucosa. Medical interventions to control hyperammonaemia have been ineffective, necessitating surgical revision of ureteric diversions. 18,20,105,107
Miscellaneous causes
Circulatory
Hyperammonaemia occurs in hypovolaemic shock and congestive cardiac failure, in which loss of hepatocytes and hence protective GS activity may contribute. Congenital portosystemic shunts are a rare cause. 10,20
Multiple myeloma and leukaemia
Hyperammonaemic encephalopathy is an uncommon complication of multiple myeloma, generally of aggressive or chemotherapy-resistant disease. In one series, overall mortality was high (44%), but was lower (32%) in those treated with chemotherapy which reduced plasma ammonia. 108 Hyperammonaemia may also occur in leukaemia, not attributable to chemotherapy. Accelerated catabolism may be contributory. 109
Chemotherapy
Hyperammonaemic encephalopathy is an uncommon, frequently fatal, complication. 110 Asparaginase hydrolyses the amide groups of asparagine and glutamine and has caused symptomatic hyperammonaemia in haematological malignancies. 111–113 5-Fluorouracil, a pyrimidine analogue which acts primarily through inactivation of thymidylate synthetase, has caused severe hyperammonaemia in patients treated for solid organ malignancies. The mechanism is unknown. The risk may be increased by infection, hypovolaemia and constipation and, possibly, polymorphisms of the thymidylate synthetase gene. 110,114,115
Anticonvulsants
Symptomatic hyperammonaemia has been induced by carbamazepine, 116 and valproate, alone or in combination with phenobarbital, topiramate or phenytoin (see above).
Salicylate
Salicylate was associated statistically with Reye's syndrome, a metabolic encephalopathy with hyperammonaemia which was reported in children under 16 y of age. 117 This led to recommendations to avoid the use of aspirin for febrile children under 12 y (USA 1982; UK 1986). In retrospect, many children with Reye's syndrome probably had inherited defects, particularly of fatty acid oxidation or the urea cycle, and the diagnosis is now made very infrequently. 118,119 The mechanisms underlying the association remain unclear. It is not shared by other NSAIDs. Salicylate inhibits β-oxidation of palmitic acid in vivo (mice) and in vitro possibly by inhibiting LCHAD, and also causes mitochondrial damage, with uncoupling of oxidative phosphorylation, inhibition of complex I of the respiratory chain and increased release of reactive oxygen species. 120–122 Because salicylate is activated to salicyl-CoA during detoxification, an alternative proposal is that it sequesters coenzyme A and thereby decreases both urea cycle activity, through NAG depletion, and fatty acid oxidation. 120
Increased protein catabolism
Ammonia production is increased by excessive amino acid catabolism, for example resulting from severe exercise, seizures, trauma or burns, steroid administration, chemotherapy, gastrointestinal haemorrhage, high protein supplementation of poorly nourished patients, arginine deficiency and bladder, uterine or joint irrigation with glycine solution during surgery. This may precipitate symptomatic hyperammonaemia in those already at risk from another cause. 15,17,47,123,124
Myxoedema coma
Rarely, patients with severe hypothyroidism and coexisting liver disease have presented with hyperammonaemic coma, without other evidence of hepatic decompensation. Symptoms resolved with thyroid replacement therapy. The mechanism is unknown. 125,126
Diagnostic tests
The key steps in diagnosis are to consider hyperammonaemia at presentation and to get an accurate plasma ammonia result quickly. A raised value must be confirmed immediately on a repeat specimen. Samples collected for metabolite analyses at acute presentation are the most informative since abnormalities may disappear rapidly after commencing treatment (Figure 4). Analysis of samples for DNA, and collection of skin and tissue biopsies require informed consent.
Diagnostic approach to severe hyperammonaemia in an adult which is not due to liver disease. CK, creatine kinase; def, deficiency; UCD, urea cycle defect; HHH syndrome, hyperammonaemia, hyperornithinaemia, homocitrullinuria syndrome; NAGS, N-acetylglutamate synthetase; CPS I, carbamyl phosphate synthetase I; AL, argininosuccinic acid lyase; FFA, free fatty acids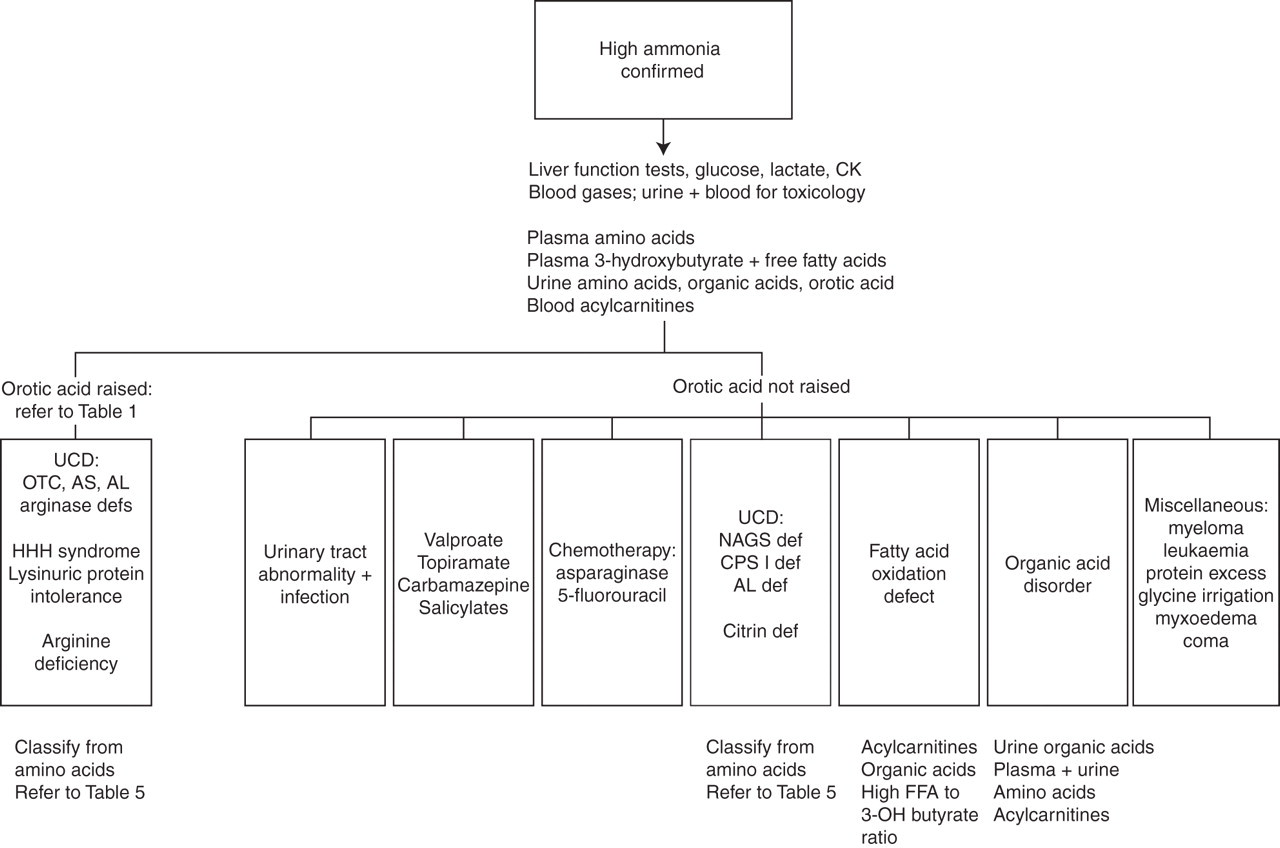
Plasma ammonia
Plasma ammonia is increased by muscle exercise, smoking, haemolysis during venesection (erythrocytes contain about three times as much ammonia as plasma 127 and adenylate deaminase which could release ammonia from adenosine monophosphate in plasma), and delayed analysis of whole blood or separation of plasma. The ammonium concentration of standing blood increases spontaneously and is a continuous process. The mean rates of increase for normal whole blood per hour were: 3.9 μmol/L at 0°C and 6 μmol/L at 22°C. Erythrocytes were the main source of increase, the contribution from plasma being 7% at 0°C and 10% at 37°C. Normal, promptly separated plasma, is stable at 4°C for four hours and for 24 h at −20°C, 128,129 but ammonia increases more rapidly in plasma from some patients, particularly those with liver disturbance or metabolic disorders. Increased deamination of amino acids by deaminases, including γ-glutamyl aminotransferase which has glutaminase activity, and/or raised plasma glutamine and asparagine are contributory. It is recommended that free-flowing blood is collected into a verified ammonia-free chilled lithium-heparin or ethylenediaminetetraacetic acid tube, avoiding use of small catheters, placed immediately on (water) ice, separated within 15 min of collection and analysed promptly. If delay is likely, plasma should be stored at −70°C. Re-freezing after thawing increases ammonia and must be avoided. Haemolysis should be documented. If not an emergency, samples taken fasting or at least six hours after food are preferable. 129,130
Most clinical laboratories analyse total ammonia using automated methods based on reductive amination of 2-oxoglutarate by glutamate dehydrogenase with generation of NAD or NADP, or dry slide microdiffusion technology in which ammonia released from the sample is detected by the change in pH of an indicator dye. Dry slide chemistry with a reflectance meter for detection is used in hand-held blood ammonia meters. Gas-sensing electrodes are also available, in which ammonia released from the sample diffuses across a gas permeable membrane into a buffer. The change in pH is detected as a change in potential between sensing and reference electrodes.
In enzymatic procedures, spuriously high results may be obtained when NADH is used for the reaction because of consumption. Preincubation to minimize this lengthens the procedure, with increasing risk of sample degradation. Because there are far fewer NADPH-consuming sources in plasma, a much shorter preincubation is needed. Hand-held meters measure whole-blood ammonia reliably within their linear range (up to 285 μmol/L) when used by trained personnel using free-flowing blood without massage. They allow exclusion of hyperammonaemia, but abnormal results must be confirmed by a quantitative method. There are concerns about their use out of hospital, and they are not suitable for monitoring hyperammonaemia when treatment decisions depend on absolute concentration. 63,129–132
Other metabolites in blood and urine 133
Blood
Lactate. Plasma concentrations rise rapidly after venesection. If not measured on a blood gas analyser, samples must be separated within 15 min. Delayed separation, haemolysis, venous obstruction with a tourniquet, problematic venesection and increased muscle activity, including seizures, cause raised levels.
Amino acids (plasma). Except in emergency, samples should be collected at least four hours after food, separated within four hours and stored at −20°C.
White cell enzymes. Seven milliliters of blood (absolute minimum 5 mL) is required to extract and wash enough cells for analysis. Whole-blood samples must be delivered to the Referral Laboratories within 24 h at room temperature and hence should not be collected on Fridays or Bank Holidays.
Urine
Amino acids, organic acids and orotic acid are analysed on random samples. If transport to the laboratory is delayed, samples should be refrigerated (4°C if overnight; −20°C if longer). Orotic acid is very stable in urine and can be analysed reliably with a variety of techniques including liquid-liquid chromatography, high-performance liquid chromatography (HPLC), gas chromatography-mass spectrometry (GC-MS), HPLC-tandem mass spectrometry (HPLC-tandem MS) and capillary electrophoresis. 14,134 Increasingly, HPLC-tandem MS is being used for metabolite analyses. It has been applied to analysis of orotic acid with other pyrimidines and their metabolites in dried urine filter paper strips 135 and there are preliminary reports of procedures for measuring orotic acid in plasma and dried blood spots 136 and for simultaneous quantification of urine arginine, citrulline, glutamine, homocitrulline, ornithine, aspartic acid and orotic acid with total sample preparation and analysis time of 20 min. 137
Conclusion
Severe hyperammonaemia has a high mortality and morbidity. When this is not due primarily to liver failure, prompt diagnosis and appropriate intervention may avert disaster. Early identification of the problem is a major challenge. If hyperammonaemia is suspected, plasma ammonia must be analysed without delay. Together with a careful history and clinical assessment, a small number of appropriate tests often provide a working diagnosis. The most useful samples are those collected at presentation. Definitive diagnosis may require specialist non-routine tests. Any patient found to have an inherited defect should wear a medical emergency bracelet, carry a card with contact details of the metabolic consultant and have a management plan for emergencies.
DECLARATIONS