
Abstract
The clinical presentation of a neonate with GRACILE-like syndrome, complex III deficiency and BCS1L mutations is discussed. This case is compared and contrasted with the original Finnish reports of GRACILE syndrome and other cases with a similar phenotype. This case confirms the pathogenicity of the BCS1L gene mutation c.166C>T, and provides support for the pathogenicity of a sequence variation, c.−588T>A, previously reported.
Introduction
Metabolic conditions that present in the neonatal period can be complex to manage and diagnose. Often these conditions are life-threatening and it is imperative to make every attempt to reach a diagnosis to guide management but also to provide information to the family of the risk of recurrence in future pregnancies. A multidisciplinary approach is required to hunt for the diagnosis. This case report presents a neonate with a rare, lethal, metabolic condition in whom the diagnosis was made with the collaboration of specialists from paediatrics, clinical biochemistry, metabolic services, genetics, nephrology and pathology.
Case report
A baby boy was born at 36 weeks of gestation to an unrelated Caucasian couple in their first pregnancy. Antenatal scans at 36 weeks showed intrauterine growth restriction, oligohydramnios and abnormal middle cerebral artery Dopplers. Birth weight was 2010 g (z-score −1.76), length 44 cm (z-score −1.83) and head circumference 30 cm (z-score −0.57). The arterial cord pH was 7.36 with lactate 3.3 mmol/L (normal range 0.5–2.0 mmol/L). He appeared to have lost subcutaneous fat but placental histology was normal. He developed hypoglycaemia and lactic acidosis with respiratory compensation on day 1 (pH 7.33, CO2 21 mmHg, HCO3 10.6 mmol/L, base excess −13 mmol/L, lactate 13.5 mmol/L). Investigations for infection were negative. A head ultrasound, echocardiogram, chest X-ray, eye examination and karyotype were normal. There were no neurological concerns.
Routine expanded newborn metabolic screening was normal but the initial clinical metabolic investigations revealed gross aminoaciduria. The urine organic acids showed lactic acidosis and a mitochondrial condition was suspected. DNA from buccal mucosa was negative for targeted mitochondrial point mutations and deletions. Glucose, potassium, phosphate, bicarbonate and albumin excretion in the urine were elevated and needed replacing, suggesting a renal Fanconi syndrome. Bicarbonate requirements were up to 30 mmmol/kg/d. Renal ultrasound and creatinine concentrations were normal.
Hypoglycaemia was problematic due to hyperinsulinism. He had an unsuppressed insulin concentration (14 pmol/L) at the time of hypoglycaemia (1.5 mmol/L) and a glucose requirement of 20 mg/kg/min, which was greatly increased from normal glucose requirements of 6–8 mg/kg/min. Blood sugars remained erratic even on diazoxide (20 mg/kg/d) with preprandial hypoglycaemia and postprandial hyperglycaemia (Figure 1). A conjugated hyperbilirubinaemia evolved (bilirubin 108 μmol/L, conjugated fraction 35 μmol/L) and severe cholestasis was confirmed on histology. Iron studies were consistent with iron overload with a raised ferritin (1174 μg/L), low plasma transferrin (0.6 g/L) and high iron saturation (100%).
Continuous interstitial blood glucose monitoring with hypoglycaemia defined as < 2.7 mmol/L. • indicates intermittent capillary blood glucose concentrations. (a) 200 mL/kg/d continuous nasogastric feeds prior to diazoxide use and (b) 220 mL/kg/d three-hourly nasogastric feeds on diazoxide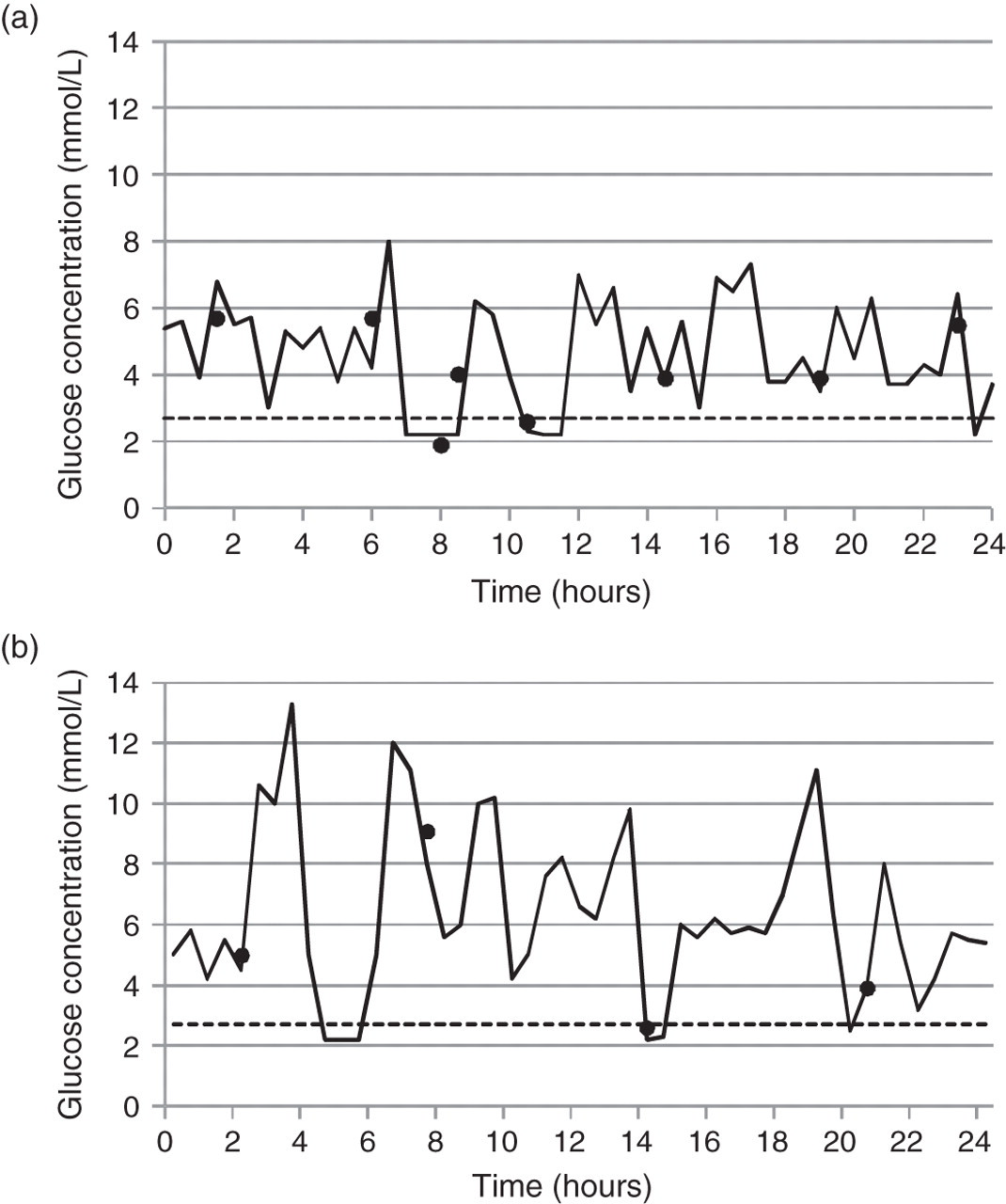
Respiratory chain enzyme assay results from skeletal muscle
Discussion
A number of pathologies can present with lactic acidosis in the neonatal period; however, persistent lactic acidosis, particularly in association with hypoglycaemia or significant hyperammonaemia, often indicates an inborn error of metabolism. In this case, other causes of lactic acidosis were excluded and further investigation for a mitochondrial condition was undertaken.
The mitochondrial respiratory chain comprises multimeric protein complexes I to V. They couple electron transport with proton transfer across the mitochondrial inner membrane in the production of ATP. 2 Complex III contains two core proteins, six small subunits and three polypeptides directly involved in electron transport (cytochromes b and c1 and the Rieske iron sulphur [FeS] protein). 1,2 All components except for cytochrome b are encoded by nuclear DNA. 2 BCS1L is a mitochondrial protein involved in the assembly of the Rieske FeS subunit into complex III. 1 More than 20 mutations in the BCS1L gene have been reported and the phenotypes vary from mild hearing loss to severe acidosis, liver dysfunction, renal tubulopathy and encephalopathy. 3 The different phenotypes probably relate to varying tissue-specific expression of the gene defect. 4
GRACILE syndrome was first described by Fellman 5 in 1998 and is caused by a mutation in the BCS1L gene. Fellman's original account included 17 Finnish children with fetal growth restriction (mean birth weight z-score −3.8), severe lactic acidosis on day 1, Fanconi-type aminoaciduria, progressive cholestasis, iron overload and all died before four months of age. Dysmorphic features, neurological signs and hypoglycaemia were not a feature. Since the original report, there have been 31 cases of GRACILE syndrome diagnosed in Finland. 3 All the Finnish cases shared the same mutation of the BSC1L gene (c.232A>G), causing a functional defect and decreased protein stability. 1,6 Respiratory chain enzymes were normal from several organs in the initial report. 5 Recently, the group further investigated complex III and discovered an assembly defect that can affect activity levels in liver, heart and kidneys but not in fibroblasts. 3 This suggests a BCS1L-dependent complex III deficiency that is tissue-specific. 3 The liver is the main target organ and severe cholestasis and fibrosis develops. Large amounts of iron are deposited in the liver when examined histologically in the first 1–2 weeks of life. However, over time, the iron overload dissipates and histological examination of the liver after one month of age often showed no signs of excess iron deposition. 7
Patients with similar clinical presentations to GRACILE patients but with different BCS1L mutations have been described in British, Turkish and Spanish children. 2,4,8,9 The cases all presented with lactic acidosis, varying degrees of growth restriction, liver and renal tubular dysfunction, encephalopathy and complex III deficiency. Hypoglycaemia was seen in the majority but not documented as being due to hyperinsulinism.
The sequence variations detected in our case were c.166C>T and c.−588T>A. The first, inherited from the father, is known to be pathogenic forming a premature stop codon. 1,4 The maternally inherited variation, c.−588T>A, is in the 5′ untranslated region and causes a single-nucleotide change in the first intron upstream from the start codon of the BCS1L gene. It has previously been reported in association with complex III deficiency and was not detected in 140 control patients of Finnish and European descent. 1,4 A pathogenic splicing event was demonstrated in this case which was not seen in controls.
A striking feature of this case was the unstable blood sugars and hyperinsulinism. Diazoxide increased the blood sugar peaks but there was still significant instability with hypoglycaemic episodes prior to feeds and rapid swings after feeding. Hyperinsulinism has been reported in patients with mitochondrial disorders and in related energy-producing conditions. It could be hypothesized that hyperinsulinism played a role in the development of lactic acidosis in this case; however, there was no discernable pattern between the blood sugar concentrations and the extent of the lactic acidosis. A screen for the cause of the severe hypoglycaemia did not initially reveal hyperinsulinism, as in the face of profound hypoglycaemia, the insulin level was suppressed. Owing to persisting hypoglycaemia, a repeat screen was undertaken one month later and this time a raised insulin concentration was found. During these two periods, the lactate concentrations were comparable. Other cases of GRACILE syndrome have reported hypoglycaemia but this was usually easily controlled and hyperinsulinism not detected. 9 All these cases still had severe lactic acidosis and so the mechanism for the acidosis is likely to be the respiratory chain complex III deficiency.
The pathophysiological basis of this disorder results from the failure of the correct assembly of complex III, in particular the incorporation of the Rieske FeS cluster, leading to defective aerobic respiration. The iron overload appears to be specific to disorders affecting the BCS1L protein and this protein may have a wider role in iron transport or biosynthesis of the FeS protein.
This case describes the first documented case in Australasia of a rare metabolic condition presenting with complex III deficiency and the GRACILE phenotype. We provide further evidence to support the significance of a previously described variation in the BSC1L gene and highlight the problematic hyperinsulinism that was unique to this case.
DECLARATIONS