
Abstract
We present an interesting case of porphria cutanea tarda with hereditary haemochromatosis and non-classical congenital adrenal hyperplasia in a postmenopausal woman. This unusual combination of conditions does not appear to be reported in the literature. The exact mechanism causing porphyria in this patient is not clear. We consider the possibility that in addition to iron accumulation and excess alcohol, altered steroid milieu may have been a possible trigger for the development of bullous lesions in this patient.
Introduction
Porphyria cutanea tarda (PCT) is characterized by decreased activity of uroporphyrinogen decarboxylase (UROD), the fifth enzyme in the pathway of haem biosynthesis (Figure 1a) and accumulation of porphyrins in the liver. It is the most common type of porphyria. These patients present with only cutaneous manifestations, which is due to photosensitivity, and have no acute neurovisceral attacks. PCT is a sporadic condition in approximately 80% of cases.
1
Some degree of iron overload is almost always present and an association between haemochromatosis (HH) and PCT is well established. Other identified risk factors include alcohol, viral infection (particularly hepatitis C), prescribed oestrogens and exposure to toxic chemicals (hexachlorobenzol and dioxins).
2
We report a case of PCT presenting together with non-classical (late onset) congenital adrenal hyperplasia (CAH) and consider the role of androgenic steroids in triggering the onset.
(a) Pathway of haem synthesis. (b) Steroid synthesis pathway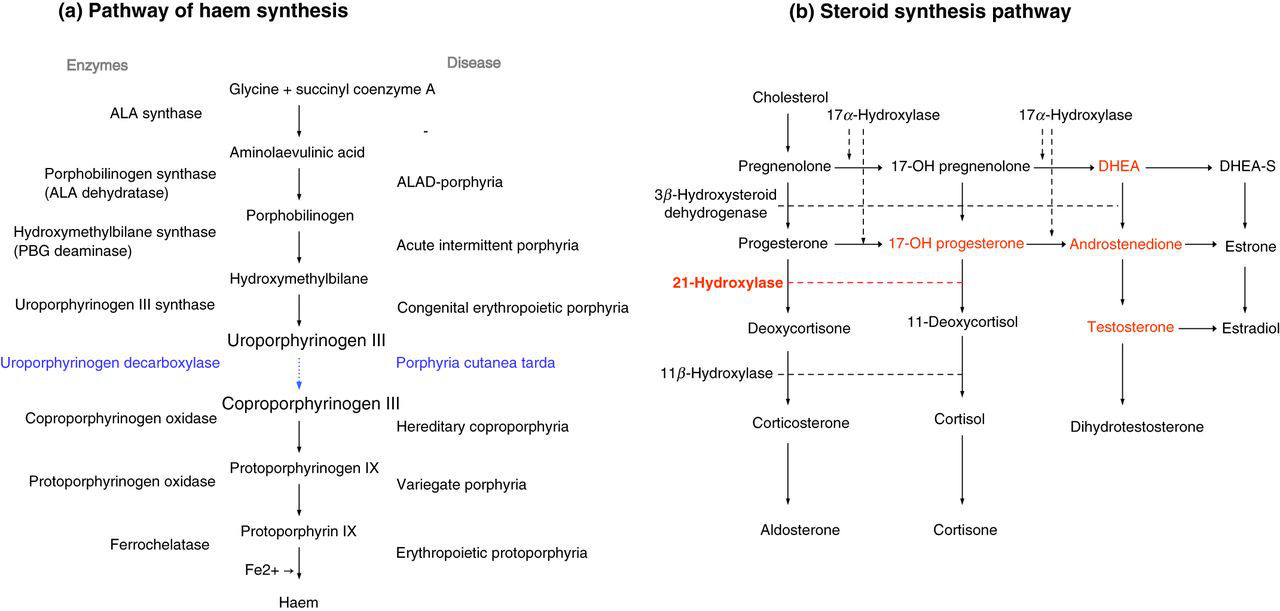
Case report
A 46-year-old postmenopausal woman presented with increased skin fragility, photosensitivity, skin pigmentation and blisters on the dorsum of both hands. She had hirsutism with excessive hair growth over her face, back, abdomen and chest (Ferriman–Gallwey score 8) and shaved daily. She smoked 25–30 cigarettes per day and consumed 24 units of alcohol a week. There was no family history of similar problems. She had been normally fertile.
PCT was clear in the differential diagnosis of her blistering skin rash, but the degree of pigmentation and hirsutism was more extreme than the facial hypertrichosis usually seen in association with this condition, prompting detailed assessment of her androgen status.
Biochemistry results at diagnosis (normal ranges in brackets)
FAI, free androgen index; DHEA-S, dehydroepiandrosterone sulphate
Clinical management
The patient was advised to avoid alcohol completely and commenced on regular venesection to control her iron stores and was also treated with low-dose hydroxychloroquine therapy (200 mg twice weekly). For her CAH, she was initially treated with dexamethasone and then switched to prednisolone, progressively reducing the dose to the minimum required to balance the need to suppress her excess androgens and avoid the side-effects of glucocorticoids.
Her blistering and skin fragility were resolved. Her pigmentation has diminished considerably, although hirsutism has remained a problem and she has gained weight. She will remain in a long-term surveillance programme for her HH.
Discussion
Sporadic PCT usually occurs on a background of chronic liver dysfunction, in which hepatic siderosis is almost universal. Hepatic UROD catalytic activity is reduced to a level that results in accumulation of uroporphyrin and carboxylated porphyrins. These hydrophilic porphyrins accumulate in the upper dermis with consequent photosensitive skin damage and the characteristic bullous skin lesions.
At a molecular level, the enzyme defect is believed to be mediated by an iron-dependent mechanism that may involve the generation of a specific inhibitor. Phillips
Alcohol excess is one of the commonest associated risk factors and our patient acknowledged regular alcohol intake above the recommended safe levels. Alcohol has a number of direct effects on enzymes in the haem biosynthetic pathway, including suppression of UROD activity and induction of ALA synthase, 4 but it may also act indirectly, causing hepatocyte membrane injury and perhaps influencing intracellular iron availability.
Our patient became symptomatic postmenopausally, with evidence of excess iron stores and abnormal liver function tests. She was homozygous for the HFE C282Y mutation that is the commonest cause of HH. Around 90% of northern European patients with HH are homozygous for the HFE C282Y mutation and a further 1–5% are compound heterozygotes for C282Y and H63D mutations.
5
While PCT is rare in HH, an increased prevalence of HFE mutations has been clearly demonstrated in PCT patient populations. Homozygosity for the HFE C282Y mutation is present in approximately 20% of USA and Northern European PCT patients.
5,6
A study of 41 British patients with sporadic PCT by Roberts
Association of PCT with non-classical CAH does not appear to have been previously reported in the literature. Miller and Levene
9
reported a case of congenital erythropoetic porphyria with CAH due to hepatic 5
In summary, we present an interesting lady with non-classical CAH, iron overload due to HH and and excess alcohol leading to the development of PCT. To our knowledge, this combination of genetic disorders in association with PCT has not previously been reported.
DECLARATIONS