
Abstract
Identification and investigation of hypoglycaemia in childhood remains an important clinical emergency. Rapid recognition and appropriate management of this clinical state continues to be important in order to prevent neurological damage or even death. The purpose of this review is to provide an update on the advances made in this area since the review by Bonham in this journal in 1993. Advances in molecular science and diagnostic techniques have assisted in understanding the mechanisms involved in the homeostasis of glucose metabolism at rest and when stressed. New disorders causing hypoglycaemia are described using the classification based upon aetiologies, which was used in Bonham's original paper. The development and use of guidelines and pre-assembled packs for investigating hypoglycaemia is also discussed.
Introduction
The identification, investigation and management of hypoglycaemia in childhood and the neonatal period remain an important clinical emergency as failure to identify this state may result in neurological damage or even death. Advances in molecular science and diagnostic techniques have assisted in understanding the mechanisms involved in the homeostasis of glucose metabolism at rest and when stressed. The purpose of this review is to update the reader on the advances made in the area since the review by Bonham in 1993. 1 Identification and molecular characterization of disease states also allow more appropriate management to be initiated and prevent unwanted side-effects.
Definition of hypoglycaemia
Hypoglycaemia can be described either biochemically or by symptoms exhibited by the patient due to the adrenergic response. However, the concentration at which symptoms are evident may be dependent on a number of factors: age of patient, pre-existing conditions, diet and maturity of the liver. It is generally accepted that at a concentration of 2.6 mmol/L symptoms are evident. The controversy that existed in the first review remains, 1–5 but some studies have shown that a concentration of <2.6 mmol/L or persistent or recurrent hypoglycaemia warrants further investigation, 2–6 particularly if risk factors are identified. For example, the requirement of a large dextrose load to maintain a normoglycaemic state or the rapid utilization of a glucose load may support the need for formal investigation of hypoglycaemia. The presence of other metabolic features such as metabolic acidosis may also warrant further studies. A metabolically compromised infant may require close supervision if their low glucose concentration (<2.0 mmol/L) persists despite feeding or if other abnormal signs develop. In the neonatal period, the classification or recognition of hypoglycaemia is particularly difficult due to the presence of alternative pathologies e.g. sepsis and therefore, for a confident diagnosis to be made, the neonate should fulfil the ‘Whipple's Triad’. 5,7 This comprises clinical manifestations, an accurately measured low plasma glucose and symptoms resolving rapidly once normoglycaemia has been re-established. During the neonatal period, significant changes occur which are closely regulated by hormonal surges to meet the energy demand of this transitional period. 5 During this stage, hepatic glycogen stores established during pregnancy maintain the infant's glucose concentrations; however, delays in feeding may allow hypoglycaemia to develop during the immediate postnatal period. 6 Stanley reports observational differences in the concentration chosen by paediatricians and paediatric endocrinologists in defining neonatal hypoglycaemia (10% <1.7 mmol/L, 66% <2.2 mmol/L, 24% <2.8 mmol/L), though he recommends a concentration of <2.8 mmol/L as a diagnostic threshold to obtain critical samples. 6 Further work is required to determine a low threshold for use in premature infants.
What is not contested is the potential neurological impairment following a symptomatic neonatal hypoglycaemic episode (see Table 1). 3,8,9 Burn et al. 9 discussed a cohort of 35 term infants who presented with symptomatic hypoglycaemia during their first five days of life collected over a 14 year period. The majority (86%) had one or more episodes of hypoglycaemia at a concentration of <1.5 mmol/L. Magnetic resonance imaging (MRI) scans were performed between 1 and 42 days of life. They observed more diverse white matter changes than previously described, and MRI was more useful in predicting severity of injury than the concentration of glucose or duration of hypoglycaemia. Growth-restricted and low birth weight infants were observed to be more prone to hypoglycaemia than controls. On follow-up at two years old, seizures and poor cognition were common, and these problems persisted through life, as reported in diabetic children following hypoglycaemic episodes. 8 The damage occurred as a result of neuronal necrosis, increased cerebral blood flow and release of neuroactive amino acids, especially aspartate that caused selective neuronal death. 8
Long-term effect of hypoglycaemia in childhood
Kindly reproduced with permission from Kanaka-Gantenbein C. Hypoglycemia in childhood: long term effects. Pediatr Endocrinol Rev 2004;
The molecular basis of glucose homeostasis and response to hypoglycaemia
In a normal fed state, glucose is obtained from the diet and any excess is stored as glycogen in the liver and as triglycerides in adipose tissue (Figure 1). Glucose homeostasis is concerned with the immediate demand for glucose which is satisfied from glycogen stores. What determines the outcome of this homeostasis is the supply of glucose to the individual and the rate of utilization of these stores. Once these stores are exhausted, glucose is provided by metabolism of alternative substrates (alanine, glycerol, free fatty acids, lactate, pyruvate), through gluconeogensis (Figure 2). The energy required for this process is provided by the oxidation of fatty acids to ketones. If the individual's endocrine response to a glucose load, increased utilization of glucose or fasting is disturbed, this may affect the individual's glucose homeostasis. Since Bonham's paper there has been an expansion in the understanding of the processes involved, particularly at the molecular level, which has allowed more appropriate treatments to be developed. 1 This review discusses the developments and advances described in the recent literature using the same classification based upon aetiologies: increased glucose utilization, impaired glycogen metabolism, impaired ketogenesis and ketone body utilization, reduced gluconeogenesis, interference with glucose homeostasis secondary to a disturbance of intermediary metabolism, other causes and associations, iatrogenic causes (drug-related hypoglycaemia), idiopathic and artefactual. 1
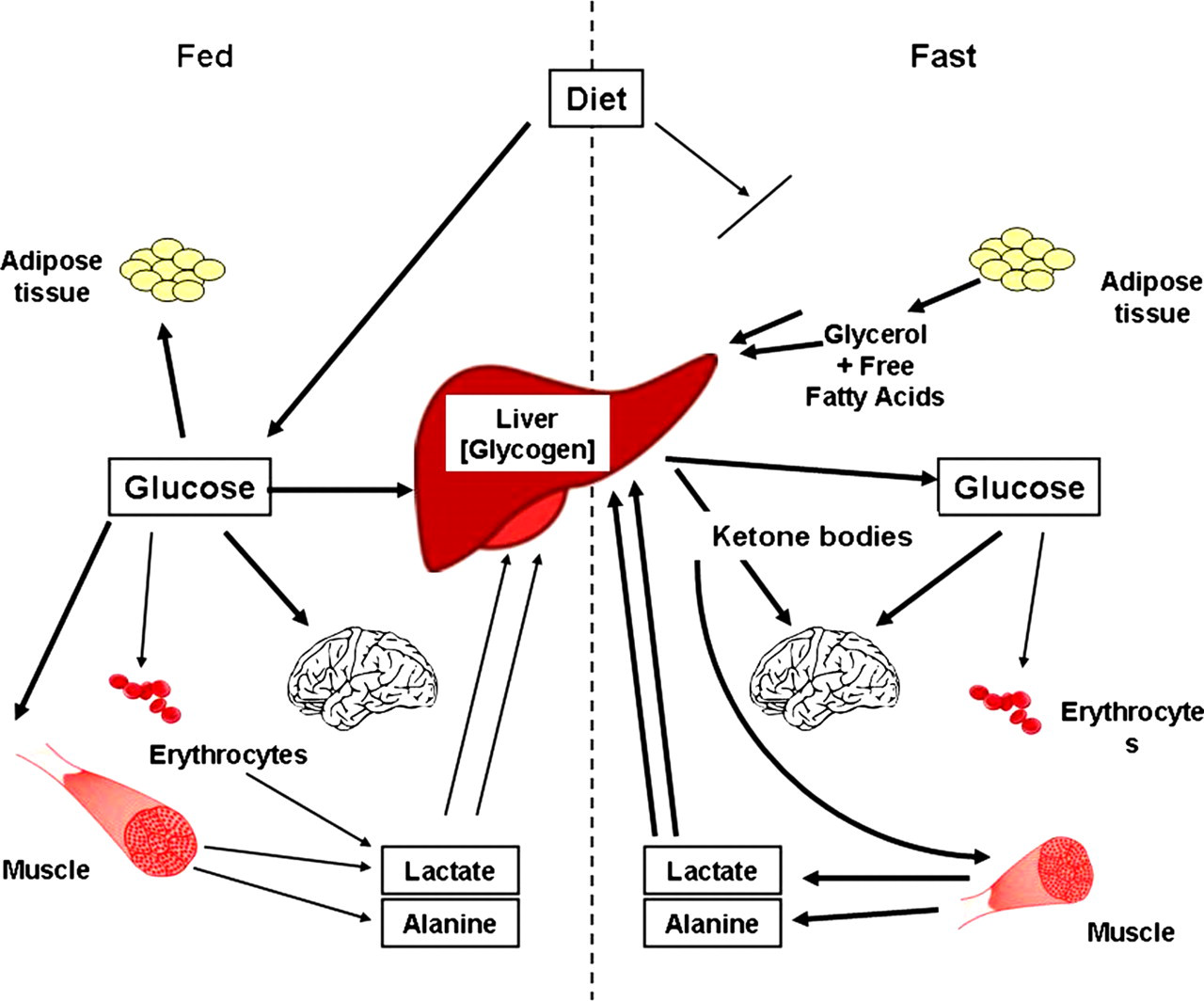
Processes involved in the maintenance of glucose homeostasis in the fed or absorptive state compared with those in the fasting or postabsorptive period. Kindly reproduced with permission of Dr J Bonham1
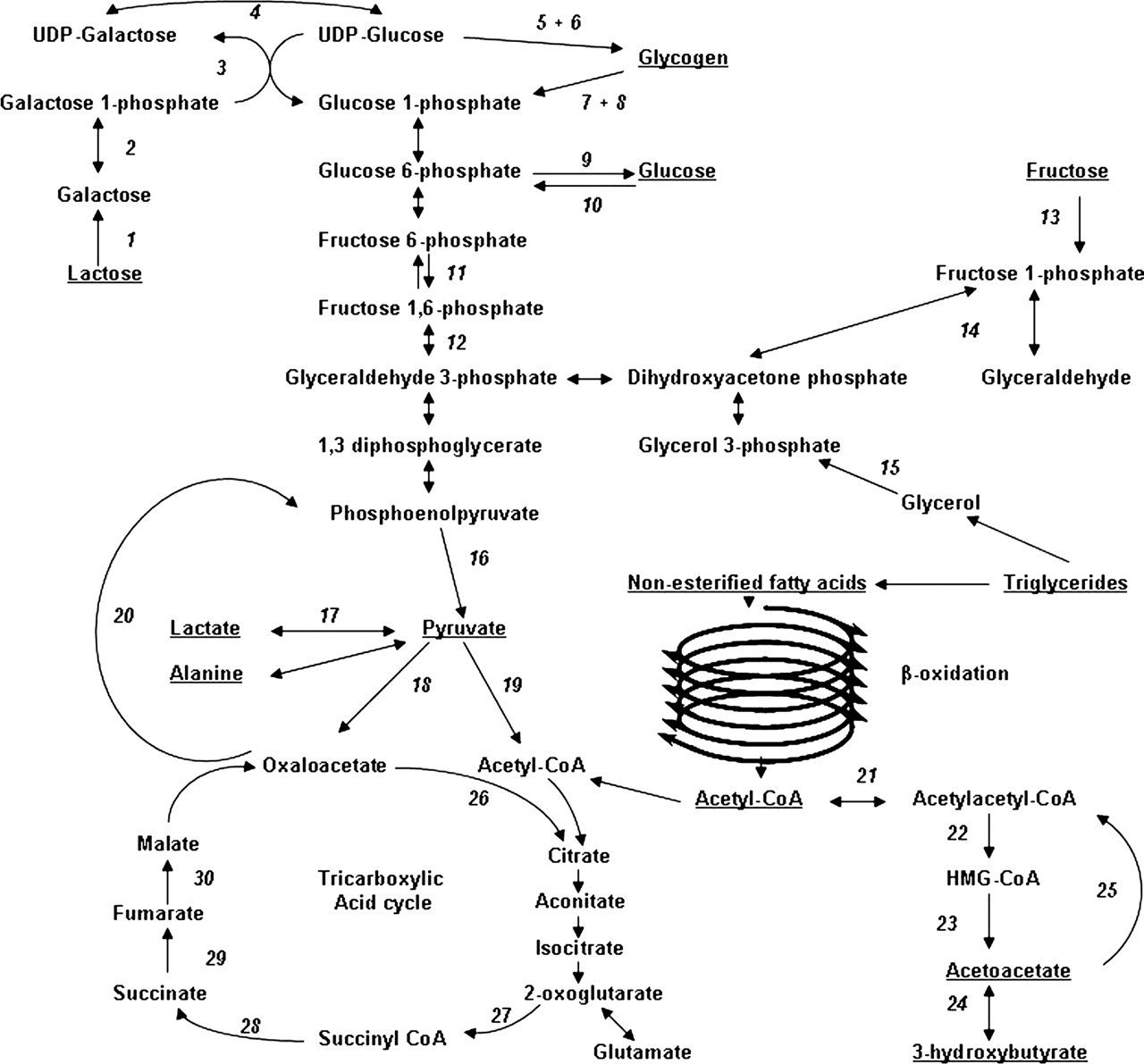
Biochemical pathways involved in glucose homeostasis. Italic numbers by arrows represent named enzymes: 1. lactase, 2. galactokinase, 3. galactose 1-phosphate uridyltransferase, 4. epimerase, 5. branching enzyme, 6. glycogen synthase, 7. phosphorylase, 8. debranching enzyme, 9. glucose 6-phosphatase, 10. glucokinase, 11. phosphofructosekinase, 12. fructose-1,6-bisphosphatase, 13. fructokinase, 14. aldolase B, 15. glycerol kinase, 16. pyruvate kinase, 17. lactate dehydrogenase, 18. pyruvate carboxylase, 19. pyruvate dehydrogenase, 20. phosphoenolpyruvate carboxylase, 21. mitochondrial acetoacetyl-CoA thiolase, 22. 3-hydroxy-3-methylglutaryl CoA synthase, 23. 3-hydroxy-3-methylglutaryl CoA lysase, 24. 3-hydroxybutyrate dehydrogenase, 25. succinyl-CoA:3-oxoacid CoA-transferase, 26. citrate synthase, 27. 2-ketoglutarate dehydrogenase complex, 28. succinyl-CoA tranferase, 29. succinate dehydrogenase, 30. fumarase
Group I: increased glucose utilization
Glucose is utilized at different rates depending on the age of subject, hyperinsulinaemia (HI) and whether the subject is compromised by a particular stress, e.g. hypoxia, respiratory distress or hypothermia. 10–13 Hypoglycaemia in the neonatal period may be more prevalent if there are associated risk factors such as low birth weight, prematurity, a smaller head size, polycythaemia, maternal anaemia and a raised maternal thyroid-stimulating hormone. 13 Therefore, if an intrauterine growth-retarded infant is delivered in an environment which makes it more prone to utilize its limited hepatic glycogen stores, an increased incidence of hypoglycaemia may be observed. 7,10,13 Fatty acid metabolism may be used as an alternative source of energy, but in the neonatal period is limited and slow due to poor muscle mass and reduced availability of substrates. However, persistent or recurrent hypoglycaemia, particularly where the individual requires a large dextrose load to maintain normoglycaemia, warrants immediate investigation in order to identify the cause, in particular HI, the commonest cause of hypoglycaemia in the neonatal population. 7,14–18 There are a number of causes that may subject the neonate or infant to an inappropriately raised insulin. The HI may be maternally driven, endogenous or exogenous in origin and the recognition of risk factors is important in the investigation and management of the patient.
Fetal insulin concentrations are determined by production and secretion by the fetal pancreas, with glucose having the main regulatory role, particularly in the third trimester where it drives growth, manages glycogen synthesis and suppresses gluconeogenesis. 10 In cases of altered concentrations of maternal glucose or delivery, growth of the fetus may be adversely affected, resulting in either macrosomia (hyperglycaemia) or a growth-restricted infant. Where there is an excess of glucose delivered, as seen in maternal diabetes or impaired glucose tolerance, increased glucose load to the fetus stimulates excess insulin secretion, which causes fat deposition. The converse is observed in low glucose states, e.g. placental insufficiency or maternal starvation, where a low glucose results in a low insulin concentration and consequently reduced growth and fat deposition (Figure 3). Once the fetus is born, insulin's primary role is glucose homeostasis and not growth. Therefore excess or inappropriately produced insulin may have serious consequences for glucose homeostasis. In the past 10 years there has been better understanding of the causes of HI, with many of the pathways being elucidated. 15 In addition, advances in diagnostic imaging techniques have radically changed the management options in the congenitally acquired form of HI. 7,15,19
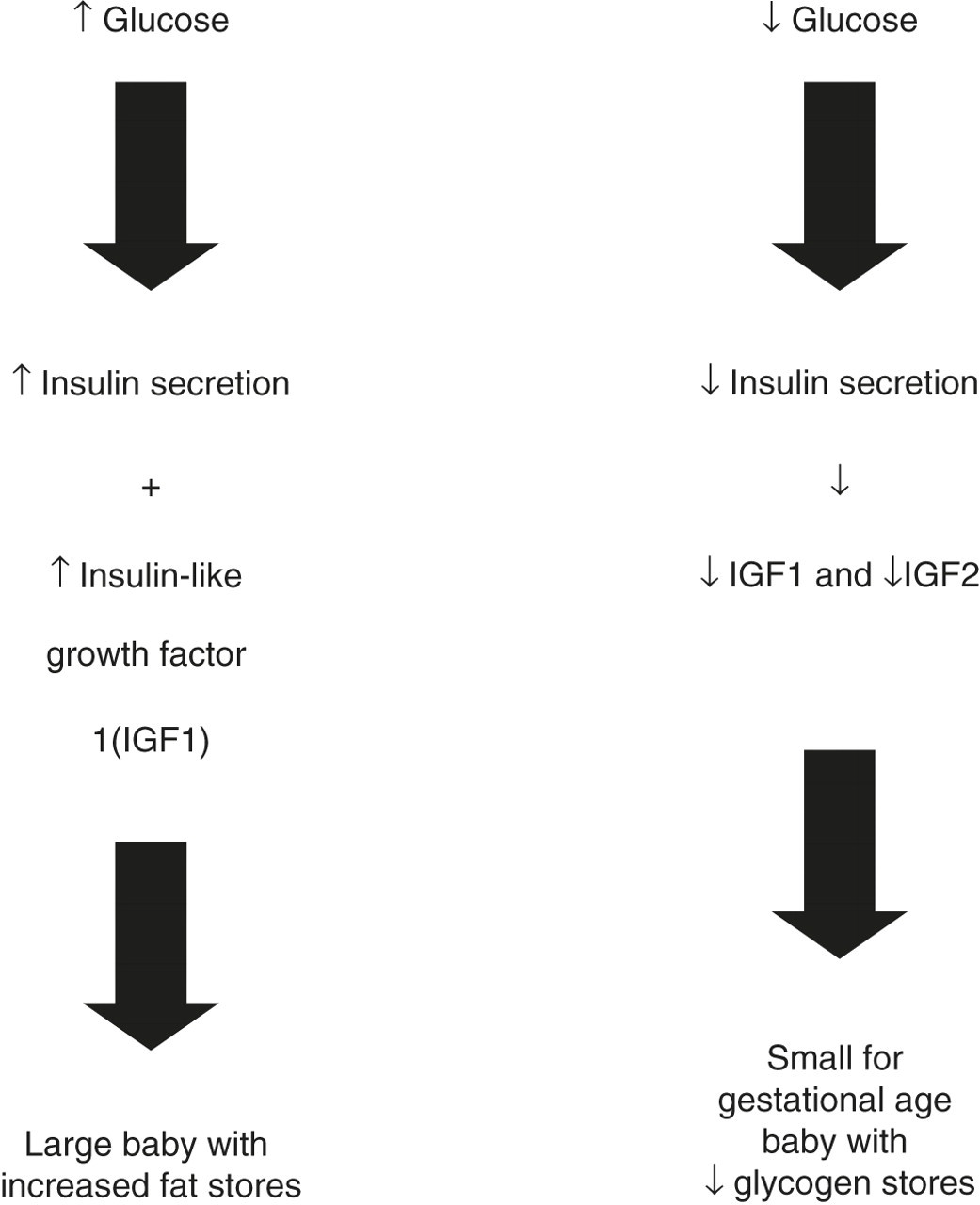
Pathway of excess and deficient glucose in prenatal period
In the presence of inappropriately raised insulin, glucose is taken up by adipose tissue and glucose production is inhibited. This also inhibits the release of free fatty acids from this tissue, restricting the substrate available for fatty acid oxidation (Randle Cycle). 20 Lipolysis and ketogenesis are suppressed, hence the characteristic finding of low fatty acids and low ketones. With no alternative energy stores for the brain, there is a significant risk of brain injury and related pathology. 15 The cause of HI may be congenital, acquired due to other risk factors or associated with developmental and metabolic syndromes (Table 2). 15 The identification of the congenital forms have allowed the elucidation of the proteins involved in pancreatic β-cell release of insulin and the action of certain drugs (discussed later). 20 Mutations in the DNA encoding the proteins may cause loss of protein function resulting in unregulated insulin release and consequently congenital hyperinsulinaemia (CHI) (Table 2).
Causes of hyperinsulinaemic hypoglycaemia
Kindly reproduced with permission from Kapoor RR, Flanagan SE, James C, et al. Hyperinsulinaemic hypoglycaemia. Arch Dis Child;
The normal response of the pancreatic β-cell to glucose uptake is an increase in the ATP/ADP ratio, which causes the closure of the KATP channel (ATP sensitive) (Figure 4). This depolarizes the cell membrane causing an influx of calcium through a voltage-gated calcium channel and exocytosis of insulin. The KATP channel is a hetero-octameric complex made up of two different subunits, and probably the most important protein in this process. There are four inward rectifying potassium subunits (Kir 6.2) encoded by the KCNJ11 gene and four sulphonylurea receptor 1 subunits (SUR1) encoded by the ABCC8 gene. 15 Loss of function results in hyperpolarization of the cell membrane and continuous insulin secretion. The drug diazoxide binds to the SUR1 receptors and acts by keeping the channel open, thereby stopping hyperpolarization (Figure 4). This is the treatment of choice in the majority of cases of CHI, apart from those patients with inactivating mutations in ABCC8 and KCNJ11 and patients with non-diffuse distribution of insulin secretion as described in focal CHI. Advances in molecular identification and imaging, particularly 18FDOPA-PET scanning, has allowed classification of patients and more appropriate pharmaceutical and surgical management. Pharmaceutical management of diazoxide insensitive CHI may be achieved using octreotide, a compound that acts on the voltage-gated calcium channel, stopping calcium influx and therefore preventing secretion by exocytosis (Figure 4). Diagnosis of CHI is made by observation of a requirement of glucose at an infusion rate >8 mg/kg/min and a laboratory measured glucose of <3 mmol/L with detectable insulin/C-peptide. However, defects in other related pathways may also result in CHI (Table 2).
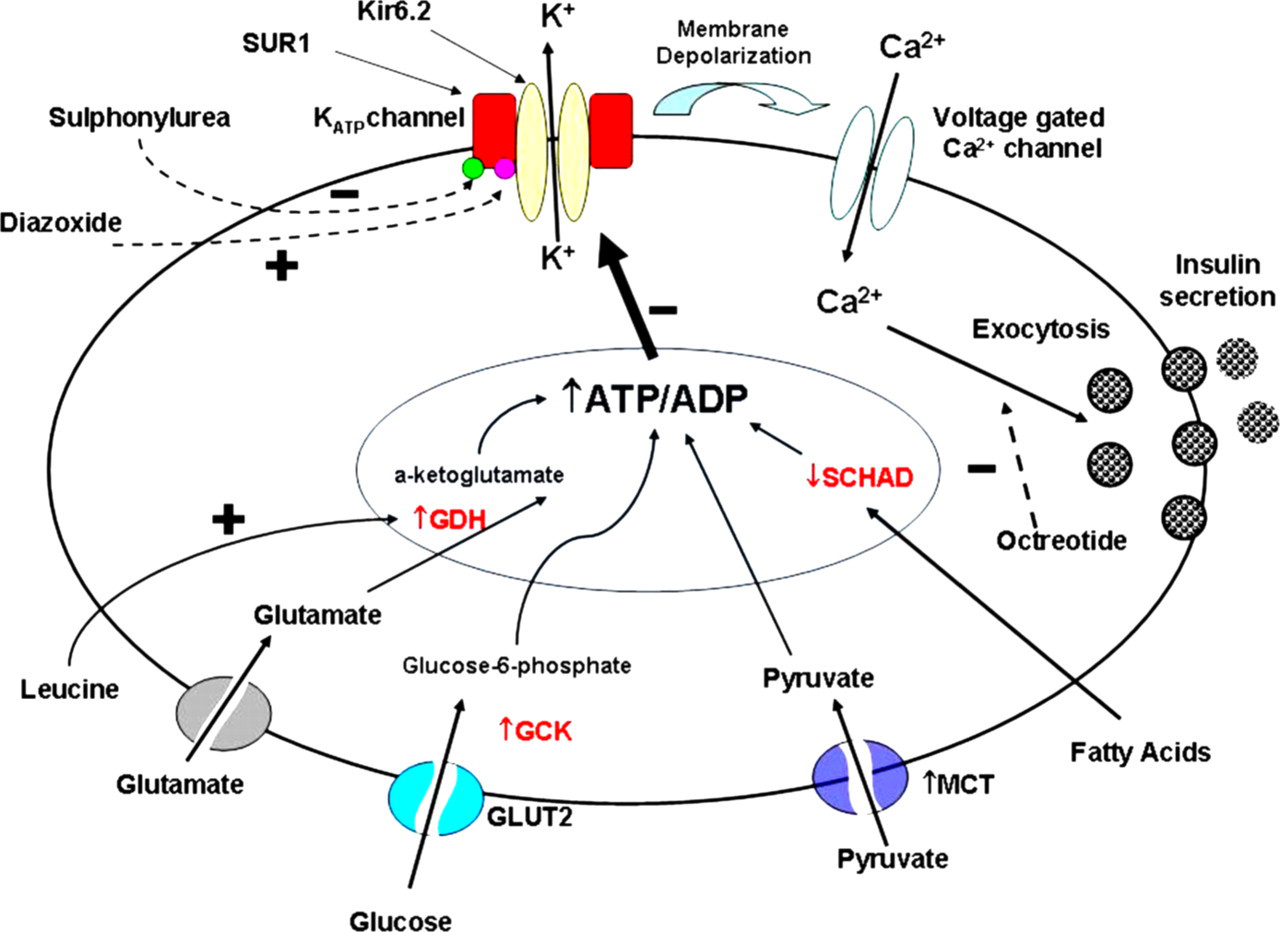
Major pathways and proteins involved in insulin secretion of the pancreatic β-cell. An increase in ATP/ADP ratio causes the closure of the KATP channel. This depolarizes the cell membrane causing an influx of calcium through a voltage-gated calcium channel and excytosis of insulin. Inward rectifying potassium subunit (yellow) (Kir 6.2) of the KATP channel, sulphonylurea receptor 1 subunit (red) (SUR1) of the KATP channel, sulphonylurea (green), diazoixde (pink), glutamate dehydrogenase (GDH), glucokinase (GCK), pyruvate/lactate transporter (MCT), glucose transporter type (GLUT2), short-chain acyl-CoA dehydrogenase (SCHAD)
Another important protein involved in the regulatory process in response to a metabolic stress is AMP-activated protein kinase (AMPK), a protein described as a nutrient sensor. This highly conserved protein evolved to respond to carbon starvation. When activated it stimulates catabolic processes including glycolysis and fatty acid oxidation and inhibits anabolic processes including gluconeogenesis, glycogen synthesis, fatty acid and protein synthesis. This protein is activated in response to an increase in the AMP:ATP ratio, as a consequence of either a reduction in ATP production or increased ATP utilization. Therefore, in response to a metabolic stress such as starvation or a low glucose where there is reduced ATP production increasing the ADP:ATP ratio and hence AMP:ATP ratio, AMPK is activated. In the pancreatic β-cell, where a different isoform of glucokinase exists having a higher K m than the normal mammalian cell isoform, the rate of ATP synthesis decreases in response to a fall in glucose, consequently activating AMPK. 21,22 Studies in skeletal muscle have demonstrated that both insulin and AMPK stimulate glucose uptake but for different roles. In addition, both also suppress enzymes involved in gluconeogenesis. 22,23
Glucokinase (GCK) catalyses the conversion of glucose to glucose-6-phosphate. Mutations have been identified in the GCK gene, which change the K m of the enzyme so that it responds to lower glucose concentrations than normal. This causes inappropriately increased insulin secretion in the presence of a low blood glucose concentration.
In the condition hyperinsulinaemia and hyperammonaemia (HIHA), patients present with protein-sensitive hypoglycaemia. This is caused by increased activity of the enzyme glutamate dehydrogenase (GDH), due to heterozygous mutations in the GLUD1 gene, causing unregulated insulin secretion by an unknown mechanism. Leucine derived from protein stimulates this enzyme through allosteric activation, catalysing the oxidation of glutamate to α-ketoglutarate and ammonia. 19 In HIHA the mutation causes an increased sensitivity to leucine, so that the enzyme is up-regulated, particularly in the presence of a high protein load or following fasting. 15,19 Patients with HIHA present with HI and persistently raised plasma ammonia concentrations.
Mutations in the pyruvate and ketone transporter (MCT1) expressed in the pancreatic β-cell results in exercise-induced HI (EIHI). Pyruvate is taken up following anaerobic respiration as a consequence of exercise and, due to mutations causing increased MCT1 activity in EIHI, this results in unregulated insulin secretion and hence severe hypoglycaemia. 15,19
Hyperinsulinaemic hypoglycaemia has been described in two forms of the ever expanding congenital disorders of glycosylation (CDG). Originally described in CDG type Ib (phosphomannose isomerase deficiency), 24,25 it has recently also been observed in patients with CDG type Ia (phosphomannose mutase deficiency). 26
Hypoglycaemia has also been described in Donohue's syndrome (leprechaunism) where there is complete or near-complete absence of insulin receptors. In this condition there are very high circulating concentrations of insulin with a corresponding low/normal concentration of insulin-like growth factor 1 (IGF1). 27 It is thought that the insulin-induced response occurs via insulin binding to liver IGF1 receptors, which are only present during fetal development and infancy. There is also a delayed clearance of insulin contributing to the persistent high concentrations. 27
Group II: impaired glycogen metabolism
Glucose obtained from the diet and not used immediately is stored in the liver as glycogen, through the process of glycogenesis (Figures 1 and 2). Reserves can be accessed when needed through the process of glycogenolysis. Deficiencies of any of the enzymes involved in these processes may result in a hypoglycaemic state due to failure to synthesize glycogen or to mobilize it. The latter results in excess hepatic storage of glycogen. Clinical manifestations of deficiencies of these enzymes have been termed glycogenoses, although they are more commonly called the liver glycogen storage disorders (GSDs). The liver-associated GSDs that present with hypoglycaemia (I, III, VI, IX) all characteristically present with hepatomegaly and short stature. Hypoglycaemia is a common finding in patients with GSD IX, due to the deficiency of hepatic phosphorylase kinase, though in this condition the clinical phenotype is highly variable. 28
A deficiency in glycogen synthetase, termed GSD0, does not result in the common clinical presentation of hepatomegaly seen in the liver GSDs, as the normal function of the enzyme is to aid the conversion of glucose to glycogen. However, patients with GSD0 still present with fasting hypoglycaemia as they are unable to rely on their diminished hepatic glycogen stores for glucose production. GSD0 is also an uncommon cause of ketotic hypoglycaemia. Gluconeogenesis and fatty acid oxidation are unaffected so hypoglycaemia following fasting is initially mild, but becomes severe on prolonged fasting. 29 Treatment consists of avoiding hypoglycaemia through frequent feeds with a high carbohydrate diet.
Group III: impaired ketogenesis and ketone body utilization
Fatty acids are metabolized through mitochondrial fatty acid β-oxidation to provide energy for gluconeogenesis. 30 As a consequence, ketone bodies are formed (ketogenesis) which may in turn be used as an energy source in all tissues. 1,31 The process of mitochondrial fatty acid β-oxidation can be split into two phases: the transport of acyl-coenzyme-A(acyl-CoA) into the mitochondria via the carnitine shuttle and secondly the β-oxidation cycle itself. Defects in either of the phases may cause the patient to present with a hypoketotic hypoglycaemia. 32–34 There are three important proteins involved in the carnitine shuttle, carnitine palmitoyl transferase I and II (CPT I and CPT II) and carnitine acylcarnitine translocase (CACT). CPT I converts acyl-CoA into acylcarnitines that can then be transferred into the mitonchondria via CACT. Once inside, CPT II lyses the acylcarnitine so that the CoA ester may enter the β-oxidation cycle. This pathway may also operate in reverse, so that any build up of a particular fatty acid as a consequence of a block in the β-oxidation cycle may be reduced by transporting out of the mitochondria and then excreted in urine or bile. This process means that identification of acylcarnitines in blood or urine assists the diagnosis of possible fatty acid β-oxidation disorders. Adequacy of carnitine supply for this process is important and any primary or secondary deficiency may affect this shuttle. Primary carnitine deficiency is caused by loss of function of the carnitine transporter, organic cation transporter 2 (OCTN2), which transports carnitine across the cell membrane. 32 Secondary deficiency may also occur due to renal loss of carnitine or inadequate supplementation of the diet.
Recently three cases of a new disorder, Acyl-CoA dehydrogenase 9 deficiency were described, two of which presented with hypoglycaemia. 35 This inner mitochondrial membrane-associated enzyme is thought to have a role in the oxidation of unsaturated fatty acids. 36 It has been observed that the loss of function of hydroxyl acyl-coenzyme A dehydrogenase (HADH), formally known as short chain 3-hydroxy acyl-coenzyme A dehydrogenase, due to a mutation in the HADH gene may cause CHI through an unknown mechanism. 15,37–39 Patients typically present in the neonatal period with severe hypoglycaemia and raised hydroxybutyrylcarnitine and urinary 3-hydroxyglutarate.
The advent of expanded newborn blood spot screening across the world using tandem mass spectrometry technology has allowed the pre-symptomatic identification of a number of fatty acid β-oxidation disorders, based on the presence of increased acylcarnitines of specific carbon chain length or a deficiency of free carnitine. 40–44 Recently, a neonatal blood spot screening programme for medium chain acyl-CoA dehydrogenase deficiency (MCADD), the commonest β-oxidation disorder, was introduced into the UK following a pilot study. 45 In the absence of screening, diagnosis of MCADD is usually in infancy or early childhood with acute metabolic decompensation precipitated by intercurrent illness and/or reduced food intake. Mortality in infancy has been described at approximately 25%. 46–48 As previously stated, the normal adaptation to fasting after utilization of glucose is to use alternative energy sources such as the metabolism of fatty acids including ketogenesis and ketone body utilization, now known as ketolysis. 1,31 Ketogenesis occurs in the hepatocytes and is highly regulated by two enzymes: 3-hydroxy-3-methylglutaryl-CoA synthase and 3-hydroxy-3-methylglutaryl-CoA lyase. Ketolysis is regulated by another two enzymes: succinyl-CoA-3-ketoacid (oxo-acid)-CoA transferase (SCOT) and mitochondrial acetoacetyl-CoA thiolase (MAT). 31 Patients with an inherited deficiency of ketogenesis may present with hypoglycaemia but with unexpectedly low or absent ketones. This is because they are unable to metabolize ketones to use as an alternative energy source. The opposite is true for disorders of ketolysis where there is a ketosis in the absence of hypoglycaemia due to a build up of excess ketones. 31
Mitochondrial respiratory chain disorders, as stated in the previous review, may affect fat oxidation and therefore cause hypoglycaemia indirectly. 1 Isolated hypoglycaemic episodes have been described in patients with complex III and IV deficiency. Mochel et al. 49 hypothesized that reduced ATP production inhibits the ATP-dependent enzymes of gluconeogenesis.
Group IV: reduced gluconeogenesis
Pyruvate is metabolized through a number of reactions to produce glucose, through the process of gluconeogenesis. Deficiencies of enzymes in this pathway, especially pyruvate carboxylase, phosphoenolpyruvate carboxylase and fructose 1,6-bisphosphatase, may result in reduced gluconeogenesis and hence a hypoglycaemic state. The action of glucagon stimulates the production of these enzymes whereas insulin inhibits their expression. The process of gluconeogenesis is also under the control of other counter-regulatory hormones including adrenaline, cortisol and growth hormone. Cortisol is released in response to hypoglycaemia and acts to increase gluconeogenesis and provision of amino acids, particularly alanine, from muscle. Growth hormone acts to stimulate glycogenolysis, lipolysis and prevention of peripheral glucose uptake. Deficiency of any of these hormones may result in a non-hyperinsulinaemic hypoglycaemic state. Deficiency of cortisol, either primary or secondary (e.g. congenital or acquired, including adrenocorticotrophic hormone [ACTH] deficiency), may result in a hypoglycaemic state and it is important to identify the cause of the deficiency. This is particularly the case in males presenting with hypoadrenalism, who should have the rare condition of X-linked adrenoleukodystrophy excluded through the measurement of plasma very long chain fatty acids.
An observation frequently made by laboratories supporting a neonatal unit is the apparent inadequate adrenal response to recurrent or prolonged hypoglycaemia. Several hypotheses have been suggested to explain this phenomenon. During the transient period following birth, when the neonate is adapting to life outside the womb, regular feeds are required to maintain normoglycaemia. However, in this period the neonate will have experienced a lower average glucose concentration, which may lower the glucose threshold at which cortisol is released. 50 Therefore, the neonate's adrenal response to hypoglycaemia may be diminished during this physiological adaptation. 50,51 The response of cortisol is also lower in infants aged less than three months and is independent of gestational age, intrauterine growth or insulin concentration but rises to normal adult concentration by six months. 50 This may explain the tendency of recurrent hypoglycaemic episodes, possibly due to the depleted cortisol. However, the recurrent episodes resolve on average by day 18 of life.
Growth hormone deficiency may present with hypoglycaemia but patients are more likely to present with growth failure or other symptoms of hypopituitarism. Growth hormone concentrations in hypoglycaemic or normoglycaemic neonates have been found to be greater than the consensus concentration of 10 ng/mL. 50 Again this is considered to be as a result of the diminished physiological response. However, the growth hormone response observed in older infants and children was poor and did not correlate with growth failure and possible pituitary failure. Combined pituitary hormone deficiency presenting in the neonatal period is also an important condition to recognize. In these patients there is neonatal cholestasis with recurrent hypoglycaemia and hypothyroidism. Early management of the hormone deficiencies results in rapid resolution of the cholestasis. 52 The role of adrenaline in regulation of glucose homeostasis is highlighted by the rare condition of dopamine beta-hydroxylase deficiency. 53 No adrenaline or noradrenaline is produced and there is primary autonomic failure. Subjects present with a complicated perinatal period including hypoglycaemia.
Group V: interference with glucose homeostasis secondary to a disturbance of intermediary metabolism
The metabolism of intermediate metabolites, for example non-esterified fatty acids (NEFAs), glycerol, acetoacetate and 3-hydroxybutyrate (3OHB), is important in maintaining glucose homeostasis. 54 Triglycerides, stored in adipose tissue, are broken down to produce NEFAs and glycerol, allowing NEFAs to enter fatty acid β-oxidation and glycerol to enter the glucogenesis pathway directly in the liver. Acetoacetate and 3OHB are then produced as a consequence of β-oxidation. As described in the previous review, 1 a number of organic acidurias, amino acidopathies and carbohydrate disorders may affect these pathways. In the organic acidurias propionic aciduria (PA) and methylmalonic aciduria (MMA), the accumulation of the propionyl-CoA ester inhibits intermediary metabolism. The ester is produced from the metabolism of branched-chain amino acids, valine, leucine and isoleucine. Defects in this pathway as seen in PA (primary or secondary due to MMA) and also in maple syrup urine disease will consequently inhibit intermediary metabolism. Patients with these conditions present with ketotic acidaemia. There have also been cases described of hypoglycaemia as a rare presenting feature of another organic aciduria. Bartlett et al. 55 and more recently Oude Luttikhuis et al. 56 both described cases of 3-methylcrotonyl-CoA carboxylase deficiency presenting with severe hypoglycaemia. This enzyme also plays a role in branched-chain amino acid metabolism. In tyrosinaemia the acute liver failure experienced by patients disrupts the liver's metabolic functions including intermediary metabolism.
The metabolism of lactose, the main carbohydrate in milk, provides an important energy source in the newborn. Lactose is metabolized to glucose and galactose, which are then used for energy production. Galactose is metabolized by a series of enzymes to produce glucose-6-phosphate, which can then either be used to make glucose or metabolized to pyruvate and lactate. In galactosaemia, the second enzyme in this pathway, galactose 1-phosphate uridyl transferase, is missing and this causes the recognizable symptoms of this condition including hypoglycaemia. Patients with Fanconi-Bickel syndrome may also present with galactosaemia. It is caused by a deficiency in the monosaccharide transporter GLUT 2 (glucose transporter 2). Patients may present with hypoglycaemia as well as with hyperglycaemia and hepato-renal glycogen accumulation. 57 In GSD type 1, the enzyme that catalyses the formation of glucose from glucose-6-phosphate, glucose-6-phosphatase, is deficient so that the glucose-6-phosphate is diverted to lactate synthesis or stored as excess glycogen.
Another defect of carbohydrate metabolism is hereditary fructose intolerance (HFI), caused by a deficiency of aldolase B. Fructose, a sugar normally found in fruit and vegetables, is metabolized in the liver to make glycogen. Initially fructose is converted to fructose-1-phosphate by fructose kinase and then converted to dihydroxyacetone phosphate (DHA-P) and glyceraldehyde (GAH) by aldolase B. DHA-P can then be used for gluconeogenesis and production of glycerol-3-phosphate. In HFI, the fructose-1-phosphate accumulates and inhibits gluconeogenesis and glycogenolysis. Glycerol kinase converts glycerol to glycerol-3-phosphate. Patients with a deficiency of glycerol kinase, or who are glycerol-intolerant via unknown mechanisms, may present with hypoglycaemia.
Group VI: other causes and associations
Bonham's paper describes a number of causes of hypoglycaemia including liver disease, gastrointestinal disease, chromosome abnormalities, neonatal associations, drug-related, malnutrition and sepsis. 1 Gastroenteritis was the commonest cause of hypoglycaemia in infancy in an audit performed on hypopacks collected. 58 It would be expected that in any condition where there was a reduction in nutritional supply due to malabsorption or malnutrition that hypoglycaemia may occur. Similarly if there is a limited store of glycogen, as in premature infants or small for gestational age neonates, then they may be more prone to hypoglycaemia as discussed previously. Sepsis was also a recognized cause of hypoglycaemia in this audit, probably due to the increased utilization of glucose. 58
There have been a number of newly recognized conditions that have been described presenting with hypoglycaemia. Søvik et al. 59 described a patient with Wolcott–Rallison syndrome who presented at 5.5 years with a Reye's-like episode with hypoketotic hypoglycaemia. Examination of urinary organic acids showed a 3-hydroxydicarboxylic aciduria, indicating a possible mitochondrial fatty acid oxidation involvement. In the neonatal form of citrin deficiency, which presents with neonatal intrahepatic cholestasis, hypoglycaemia has been observed. The defective gene, SLC25A13, encodes for an aspartate-glutamate carrier protein called citrin, which has been hypothesized to provide a substrate for gluconeogenesis. 60
Group VII: iatrogenic causes (drug-related hypoglycaemia)
Where the first review discussed preoperative starvation and renal transplants causing hypoglycaemia, this review will focus on a number of drugs that are associated with hypoglycaemia, which were discussed in a recent systematic review. 61 This review found that there was a weak association between hypoglycaemia and a number of drugs not used in the management of diabetes. Those that had a low to moderate evidence base included the quinolones, pentamidine, quinine, beta-blockers, angiotensin-coverting enzyme inhibitors and insulin-like growth factor. The majority of patients discussed in this paper were elderly. However, some of the listed drugs may occasionally be used in the management of paediatric patients, for example indomethacin in the treatment of patent ductus arteriosus. Those patients who did present were considered severe if they were symptomatic and/or required another person's assistance. Of more importance is the awareness of the misuse of drugs used in the management of diabetes or of insulin administered accidentally or maliciously. Sulphonylureas bind to receptors on the KATP channel and stimulate insulin secretion, as described previously (Figure 4). There are a number of drugs available, with the second-generation drugs being more potent than those used previously. The age of exposure is wide (6 months to 14 years) and ingestion may be accidental or deliberate. 62 Administration of exogenous insulin should also be suspected in a child presenting with hypoglycaemia, especially if severe, and has been implicated in several high-profile attempted or actual homicides. Munchausen by proxy or self-poisoning must be considered in the differential diagnosis of a previously healthy child presenting with hyperinsulinaemic hypoglycaemia, particularly as the consequences may be significant if not detected. 63 The availability of diabetic drugs and insulin in the child's environment and use by family members or carers must be questioned. Access to other drugs, chemicals and alcohols should also be ascertained in suspected poisoning cases. A simple strategy in suspected exogenous insulin administration is to simultaneously measure insulin and C-peptide at the time of hypoglycaemia. In exogenous insulin use there will be no C-peptide produced, whereas in an insulinoma there will be a corresponding rise in C-peptide concentrations. However, it is important that the clinicians and laboratory staff are fully aware of the limitations of the current commercial insulin assays for the detection of recombinant insulin analogues. 64,65 There is significant cross-reactivity with most available recombinant insulin preparations. 64–66 Suspected sulphonylurea ingestion may be investigated by identification of these drugs in the patient's plasma or urine.
Group VIII: idiopathic
Children who show an exaggerated physiological response to hypoglycaemia have been described as having idiopathic ketotic hypoglycaemia (IKH). 67,68 Children tend to present with IKH between the ages of 18 months and seven years, 58 the commonest time when hypoglycaemic episodes are observed. 16,17 The exact pathogenesis of IKH is unknown; however, Bodamer et al. 68 have shown using isotope studies that it may be due to a failure to maintain hepatic glucose production and not increased glucose oxidation. More recently Huidekoper et al. 67 have supported this view and hypothesized that IKH may represent ‘the lower tail of the Gaussian distribution of fasting intolerance in children’. IKH may be more pronounced in younger infants due to the limited stores and higher glucose utilization. 11,12,67 This condition normally disappears by age 6–7 years, 68 as glycogen stores and fasting intolerance improves, supporting the hypothesis that it is a physiological not a pathological state.
Group IX: artefactual
Group IX is a useful addition to the aetiology-based classification, which describes the method-related causes of hypoglycaemia. The limitations of analytical methods used to measure glucose concentrations need to be recognized. In particular, point of care (POCT) assays may perform poorly at low concentrations of glucose, giving falsely low or high readings, a fact which has been highlighted by a number of Medical Devices Agency (MDA) alerts and papers. 69–74 All POCT measurements should be performed by appropriately trained staff following the relevant standard operating procedure. The need for awareness of false-negative and false-positive readings must be highlighted especially to the clinical users, 73 as under- or overtreatment may result in permanent neurological damage. Situations where significant limitations of the use of POCT exist include severe dehydration caused by diabetic ketoacidosis, hypotension, shock, peripheral vascular disease and hypertriglyceridaemia/lipaemia. A number of physiological phenomena are also known to affect POCT glucose measurement, particularly in the newborn where anaemia falsely increases and polycythaemia falsely decreases the result. 73 Ideally, POCT meters should have good precision at low glucose concentrations but should not replace the laboratory-based assay. 75 In addition to over- or underestimation by glucose meters, the user should also be aware of spurious causes of hypoglycaemia due to analytical effects or misuse of the meter. 72,74 In addition, the MDA has recommended that some types of glucometers should not be used in certain patient groups, particularly neonates, due to performance issues. 14
Laboratory investigation of hypoglycaemia
The investigation of symptomatic hypoglycaemia is a clinical emergency. In the last 10 years there have been numerous guidelines developed for the biochemical investigation of hypoglycaemia including the best practice guidelines of the MetBioNet (National Metabolic Biochemistry network) (
If it is not possible to obtain the appropriate fasted samples or they are not collected at all, the clinicians may decide that a deliberate fast is required to precipitate hypoglycaemia. However, the procedure is not without risk and should therefore only be carried out in an experienced unit with the appropriate guidelines and in appropriately selected patients. 76
The repertoire of investigations suggested by guidelines and contained in packs should be evidence-based and supported by an appropriate laboratory service providing analysis urgently within an agreed turnaround time. 77 It is the responsibility of the referring laboratory that samples are sent to a referral laboratory providing this level of service. It is also very important that contact details of the referral laboratory or specialist clinician are available to the local clinicians and laboratory staff using the protocol and packs to enable them to obtain specialist advice. This is particularly important when there is a limited volume of blood available, which must be prioritized in order to obtain the most appropriate information for initial management and diagnosis. This is not an uncommon problem so liaison between laboratories and clinicians is vital. Table 3 describes possible clinical symptoms and prioritization of samples. If necessary, some investigations, highlighted in Table 3, may be collected following initial correction of hypoglycaemia, if not collected when hypoglycaemic. There are several diagnoses discussed in the text which may be rare (see Table 4 for prevalence of possible conditions) and may not be identified using the investigations contained in a hypopack. For these, discussion with the referral laboratory or specialist metabolic clinician is essential so that the appropriate investigations are performed. However, the investigations performed in a hypoglycaemia screen should in the first instances assist with the recognition and management of a hypoglycaemic episode and identify the majority of the common and/or treatable causes of hypoglycaemia.
Laboratory investigations for suspected hypoglycaemia and specific clinical symptoms
*Samples that may be collected postcorrection of hypoglycaemia
Relative frequencies of conditions that may present with symptoms of hypoglycaemia
It is important that clear guidance is given in such protocols of when the packs should be used, whether on the basis of clinical symptoms or of POCT blood glucose measurement. When a patient with symptoms of hypoglycaemia is admitted to an emergency department it is common practice to perform a urine dipstick test for ketones. However, the guidelines should highlight to the user that a fatty acid oxidation defect is possible despite the presence of ketones (urine or blood). 84,85 Therefore it is essential that appropriate samples are collected to exclude this, even if a patient has ketones on a urine dip stick. However, it is important to remember that investigations are only one part of an effective work-up of an acutely ill child and a full clinical history, family history and examination are important too.
Recently, there has been discussion in the literature concerning the inclusion of growth hormone and cortisol in such guidelines, and whether they are specific enough to confirm an isolated growth hormone deficiency or adrenal insufficiency. 86,87 One study reviewed growth hormone and cortisol concentrations measured in patients deliberately fasted as part of a controlled test. 86 They found that, in those patients where growth hormone deficiency had not been excluded initially, that the specificity was only 30% when using a concentration of <7.5 ng/mL. They re-iterated that the child's growth should also be considered when interpreting a growth hormone result. In the neonatal period, there is probably no justification for measuring the growth hormone concentration as studies have shown poor discrimination between hypoglycaemic and normoglycaemic subsets. 50 Those patients with suspected Addison's disease were also less likely to present with hypoglycaemia. However, personal practice has shown that may not be the case. 16,17 It is also important to note the exclusion of glucocorticoid use in suspected patients. Others have taken a more robust view and removed growth hormone from the protocols entirely. Elder et al., 87 following a prospective study, removed growth hormone from their guidelines and argued that cortisol should also be removed.
Conclusion
Hypoglycaemia in infancy continues to be an important clinical emergency, and early recognition and treatment are paramount to ensure a good prognosis. Recent advances in the molecular field have allowed a better understanding of the proteins involved in glucose metabolism from conception through to adulthood. This has also allowed better drugs to be developed for certain conditions. Of particular note is the change in the approach to managing CHI. Advancements in radio-imaging, molecular tools and surgical techniques have radically changed the management of this condition. Extended neonatal screening programmes now exist in a number of countries for various conditions that may present with hypoglycaemia, particularly MCADD in the UK. However, it is still important to identify those patients not diagnosed through a programme and ensure the procedures and emergency regimens are in place to assist in the management of such conditions (e.g. British Inherited Metabolic Disease Group emergency regimes,
The role of the laboratory continues to be important in the investigation of hypoglycaemia, both in the provision of an analytical service, and in the production of guidelines or hypopacks. Throughout this process it should be remembered that the laboratory's role is only one piece of the jigsaw and should be a partnership between clinicians and laboratory staff.
DECLARATIONS