
Abstract
Background
Neutrophilic polymorphonuclear leukocytes play a crucial role in the host defence against bacterial and fungal infections. They participate in the inflammatory response through the liberation of peptides and enzymes like myeloperoxidase (MPO). Therefore, MPO has a potential as a marker enzyme for the diagnosis of wound infection.
Methods
Substrate specificities and reaction pathways of MPO were investigated for new MPO substrates: crystal violet, leuco crystal violet, fast blue RR (4-benzoylamino-2,5-dimethoxybenzenediazonium chloride hemi(zinc chloride) salt) and various systematically substituted model substrates based on 2,7-dihydroxy-1-(4-hydroxyphenylazo)naphtalene-3,6-disulphonic acid. In addition, fast blue RR was covalently bound to siloxanes allowing immobilization of the substrate, while cellobiosedehydrogenase was integrated for generation of hydrogen peroxide required by MPO.
Results
Elevated concentrations of MPO were found in infected wounds compared with non-infected wounds (92.2 ± 45.0 versus 1.9 ± 1.8 U/mL). Various soluble and immobilized substrates were oxidized by MPO in wound samples and the influence of substrate structure and reaction pathways were elucidated for selected compounds.
Conclusions
Incubation of different MPO substrates with infected wound fluid samples resulted in a clear colour change in the case of elevated MPO concentrations, thus allowing early diagnosis of wound infection.
Introduction
Myeloperoxidase (MPO) is a haeme-containing protein, consisting of two dimers connected by a disulphide bond. Each dimer comprises a β-heavy subunit (59 kDa) and a light α-subunit (14 kDa). 1 MPO is stored in the azurophilic granules of neutrophils and in the lysosomes of monocytes 2 and is not released until activation and degranulation of neutrophils. 3 MPO catalyses the production of hypochlorous acid by converting hydrogen peroxide and chloride ions. 4 Hypochlorous acid is the most powerful bactericidal oxidant produced by neutrophils. 5
Neutrophils are normally the first cells at the site of inflammation or injury. 6 The infiltration of neutrophils to injured tissue is a characteristic tool of the host defence and inflammation, 7 as neutrophilic polymorphonuclear leukocytes (PMNs) play a crucial role in the host defence against bacterial and fungal infections. They participate in the inflammatory response through the liberation of peptides and enzymes like MPO. 8 Leukocyte activation prompts the release of MPO and the formation of oxidants important in host defence. 9 Therefore, elevated concentrations of MPO are a potentially useful marker for quantification of tissue PMN accumulation. This is correlated with MPO release by neutrophils due to inflammatory processes.10,11
MPO concentrations in blood are now considered as a marker of neutrophil activation and degranulation.12,13 However, measurements by enzyme immunoassays quantify the concentration of the enzyme without measuring its true enzymatic activity. Nevertheless, different studies have shown the importance of MPO for cardiovascular diseases like coronary artery disease, acute coronary syndrome and acute myocardial infarction.8,14 As a consequence, elevated MPO concentrations can be used for predicting future cardiovascular events, after controlling for C-reactive protein (CRP) and other risk factors. 15
Additionally, concentrations of MPO are significantly higher in the tissues of pressure ulcers compared with those of acute healing wounds. 16 It is widely assumed that wound fluid (WF) has the potential to provide important biochemical information. This information can be used as a diagnostic indicator providing information for determining the overall status of a wound and for monitoring the progression of wound healing. Furthermore, WF can be used to monitor specific parameters that have been identified as prognostic indicators or are targets of a particular therapy.17–19
Wound infection is a severe complication during wound healing causing diagnostic and therapeutic problems. Currently, detection of wound infection is based on evaluation of the well-known signs of inflammation like rubor (redness), calor (heat), tumour (swelling) and dolor (pain) by medical doctors and/or time-consuming procedures requiring special machinery like identification of the causative organism(s) after wound swabbing or determination of serum CRP. However, both methods are not suitable for rapid diagnosis of wound infection.20–22
As there is a strong need for a fast prognostic aid that would assist in predicting clinical infection of a wound prior to obvious clinical symptoms, we have determined MPO activity in different types of chronic wounds. Based on these data, we have investigated enzyme responsive devices using novel MPO substrates for simple detection of this enzyme and, as a consequence, the development of a rapid and simple diagnostic system for detection of wound infection.
Materials and methods
Sample collection and preparation
Fluids from eight postoperative wounds, three decubitus ulcer wounds, three malum perforens wounds and three ulcus cruris wounds were collected and analysed by biochemical techniques. Exudates were collected directly from the exudating wound surface by using needles or a spoon before the daily treatment. Four samples were collected during Vacuum Assisted Closure Therapy (negative pressure wound therapy system) in special canisters without starch. Additionally, fluid from 10 blisters was collected. The samples were centrifuged three times at 420 g for 20 min to remove cells and tissue material and stored at −20℃ until analysed. Eight of the 17 wounds were described as clinically infected by the attending doctors. The patients studied so far were grouped and labelled with the following abbreviations (followed by a number): i = infected wound; n.i. = non-infected wound; b = blisters.
Determination of haemoglobin concentration
The haemoglobin concentration in the samples was determined spectrophotometrically at 405 nm using haemoglobin as a standard (0.5 μg/mL to 2.5 mg/mL haemoglobin human [Sigma-Aldrich, St Louis, MO, USA]). In this concentration range, eventual oxidation of MPO substrates by haemoglobin was recorded and subtracted from MPO activity data given below.
Determination of MPO activity
MPO activity was determined according to the method described by Desser et al. 23 Briefly, 10 μL WF or enzyme solution was mixed with 290 μL substrate solution, containing 100 mmol/L Guaiacol (Sigma-Aldrich) and 0.017% (w/w) H2O2 in 50 mmol/L potassium phosphate buffer (pH 7.0). Enzyme standards containing 0.16 U/mL up to 10.0 U/mL MPO (Sigma-Aldrich) were prepared for calibration. The formation of tetra-guaiacol was measured every 20 s for 15 min at 470 nm. The change of optical density per minute was calculated from the initial rate. Additionally, the increase in absorbance after 100 s was used for calculation of MPO activity. The measurements were repeated five times.
Different MPO substrates
TMB and ABTS
As previously described by Bozeman et al., 24 TMB (3,3′,5,5′-tetramethyl-benzidine) and ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid)) were used for assaying MPO activity. Therefore, 3346 μL of succinate buffer (pH 5.4) comprising 0.3 mol/L sucrose were used, and 105 μL 1% (v/v) H2O2 and 49 μL of the substrates (10 mmol/L ABTS or 25 mmol/L TMB) were added. TMB was firstly dissolved in N,N-methylformamide, while ABTS was dissolved in water. Five microlitres of infected and non-infected WF samples were incubated with 100 μL of the substrate solutions for the detection of MPO activity. Visual inspection of the samples after 30 min of incubation at room temperature was carried out. Additionally, absorbance was measured at 410 nm (ABTS) and 650 nm (TMB). The experiments were repeated five times and the coefficient of variation was determined.
Fast blue RR
Fast blue RR (4-benzoylamino-2,5-dimethoxybenzenediazonium chloride hemi(zinc chloride) salt) was tested for its ability to be used as the MPO substrate. The reaction mixture contained 5 mL sodium acetate buffer (200 mmol/L, pH 4.0) and 500 μL fast blue RR (0.5 mmol/L in 0.06 M HCl). Eight microlitres of H2O2 were added to 2 mL of reaction solution. For initiation of the colour-reaction, 5 μL of sample (MPO or WF samples) was mixed with 295 μL of the reaction mix. Visual inspection of the samples after 30 min of incubation at room temperature was carried out. Additionally, absorbance was measured at 305 nm. The experiments were repeated five times and the coefficient of variation was determined.
Leuco crystal violet
The leuco crystal violet (LCV) method is based on the oxidation of 4,4′,4″-methylidynetris (N,N-dimethylaniline, LCV) in the presence of H2O2 and peroxidase, forming a crystal violet ion (CV+) which has an absorbance maximum at a wavelength of 590 nm. CV+ is stable for several days. 25 Oxidation of LCV to crystal violet was carried out using an assay modified from the previously published method from Pricelius et al. 26 The reaction mixture contained 5 mL sodium acetate buffer (200 mmol/L, pH 4.0) and 500 μL LCV (1 mmol/L in 0.06 M HCl). To 2 mL of reaction solution, 8 μL H2O2 were added. To initiate the colour reaction, 25 μL MPO or 5 μL WF samples were mixed with 275 or 295 μL of the reaction mix. Visual inspection of the samples after 30 min of incubation at room temperature was carried out. The experiments were repeated five times, and absorbance was measured at 590 nm.
Crystal violet (Gentian violet)
An aqueous dye solution (1% mg/mL) was prepared. To assure total dissolving of the dye, the solution was incubated for 15 min with ultrasonic radiation. Then, 70 μL of this solution were diluted in 10 mL potassium phosphate buffer (100 mmol/L, pH 7.0). To 2 mL of this reaction mix, 8 μL H2O2 (1% v/v) were added. For measuring the enzyme conversion, 100 μL dye solution and 25 μL MPO (150 U/mL) were used. Additionally, enzyme conversion was carried out using infected and non-infected WF samples (data not shown). Visual inspection of the samples after 60 min of incubation at room temperature was carried out. Absorbance was measured between 400 and 800 nm.
The conversion of crystal violet by MPO was measured with high-performance liquid chromatography/mass spectrometry (HPLC/MS). The MS spectra were acquired with an Agilent Ion Trap SL (Agilent Technologies, Palo Alto, CA, USA) equipped with an electrospray ionisation operated in positive and negative ion mode and the electrospray voltage was set to 3500 V. Dry gas flow was set to 10 Lmin−1 with a temperature of 350℃, nebulizer set to 50 psi. As eluent, a mixture of acetonitrile/formic acid/water (80/8/12 %v/v) was used with a flow rate of 0.8 mL/min for 30 min. Scans were carried out from m/z 100–1000.
For analysis of the degradation products, 70 μL of an aqueous crystal violet solution (1% [w/v]) was diluted in 10 mL ammonium acetate buffer (pH 6.8). Buffer, MPO (150 U/mL) and H2O2 (1% [w/v]) were incubated together with the dye solution. Incubation was carried out at 37℃ with a relative humidity of 70% for 24 h. After 0, 1, 6 and 24 h, reactions were stopped by cooling down the solutions to −72℃. After cooling, the solutions were transferred into glass vials that were placed in the auto-sampler of the liquid chromatography (LC) system for automation of the MS injection.
Systematically substituted azo dyes for the detection of MPO-activity
Eighteen structurally related azo dyes with 2,7-dihydroxy-1-phenylazonaphtalene-3,6-disulphonic acid as basic structure were synthesized as previously described27–33 (Figure 1). These dyes had been systematically substituted with a representative set of functional groups at the benzene ring in ortho-, meta- or para-position to the chromophoric centre to test the stearic influence of different substitution patterns.
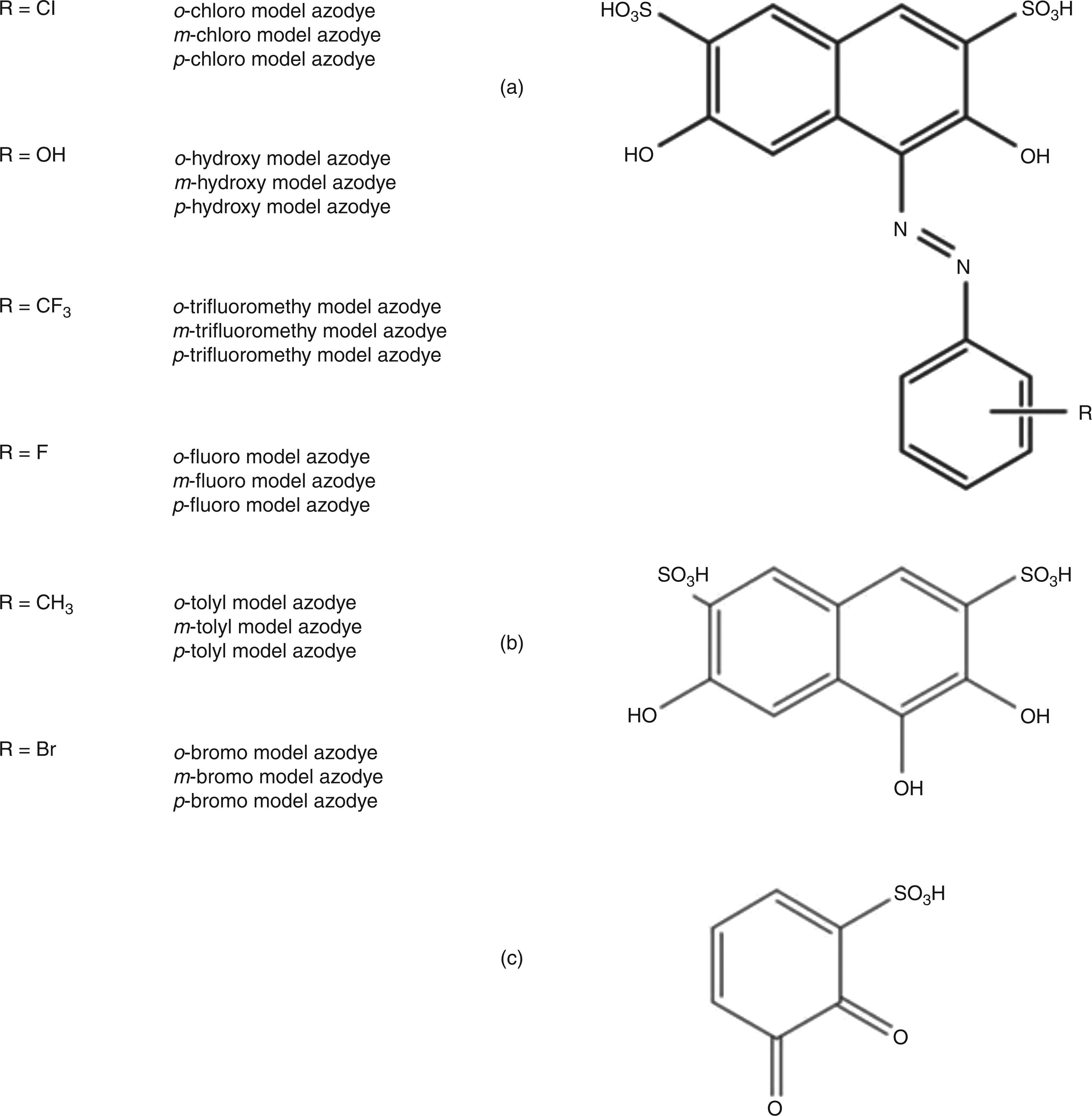
Molecule framework (2,7-dihydroxy-1-phenylazonaphtalene-3,6-disulphonic acid) of synthesized azo dyes (a) and possible oxidation products (b, c)
The reaction mixture contained 5 mL sodium acetate buffer (200 mmol/L, pH 4.0) and 500 μL model dye (5.0, 1.0 and 0.5 mmol/L in 0.06 M HCl). To 2 mL of reaction solution, 8 μL H2O2 were added. To initiate the colour reaction, 5 μL of MPO (350 U/mL) (data not shown) or WF samples were mixed with 295 μL of the reaction mix. Absorbance was measured between 400 and 800 nm after 30 min of incubation.
For HPLC separation of the oxidation products of these substrates, a Waters C8 column (Waters Corporation, Milford, MA, USA; 3.5 μm) of dimensions 2.1 mm i.d. (inner diameters) × 50 mm was used with an eluent consisting of a mixture of 90 parts acetic acid (0.1%) and 10 parts methanol (95%) containing 0.1% acetic acid. The MS spectra were recorded with an Agilent Ion Trap SL; electrospray voltage was set to 4000 V. Dry gas flow was set to 8 L min−1 with a temperature of 350℃, nebulizer set to 40 psi. Scans were carried out from m/z 50–500.
Immobilization of MPO substrates
Coupling of substrates to alkoxysilanes
Crystal violet, 2,7-dihydroxy-1-(4-hydroxyphenylazo)naphtalene-3,6-disulphonic acid and fast blue RR (N-(4-amino-2,5-dimethoxyphenyl) benzamide) were assessed for coupling to alkoxysilanes for immobilization. 2.67 g of fast blue RR were dissolved in 75 mL of absolute tetrahydrofuran in a 100-mL three-neck round-bottomed flask. With a syringe, 2.51 g of 3-(triethoxysilyl)propyl isocyanate (Sigma-Aldrich) was slowly added. The reaction mixture was then heated to reflux and maintained at this temperature for 29 h. The solvent was removed on a rotary evaporator and the residue was suspended in ethyl acetate. The organic phase was extracted three times with 0.01 mol/L hydrochloric acid and the combined aqueous phases were solvent extracted with ethyl acetate. The crude product (5.10 g) was re-crystallized from petrol ether:ethyl acetate (1:1), resulting in 3.15 g (65%) brown acicular crystals.
The molecule structure of the synthesized substrate was proven by 1H and 13C nuclear magnetic resonance (NMR) as is shown for fast blue in Table 1. In addition, successful coupling was verified with Fourier transform infrared spectroscopy (FT-IR) measurements (Bruker Tensor 35; Ettlingen, Germany). Significant changes within the FT-IR spectra of fast blue RR and the purified coupling product were observed by attenuated total reflectance (single reflection diamond cell; 32 scans). The amine group signal of fast blue RR (νs 3434 cm−1, νas 3355 cm−1) disappeared in the coupling product and a broad amide signal appeared at 3303 cm−1. In addition, a new IR band was measured at 1075 cm−1, representing the signals of the introduced alkoxysilane substituent (ν (SiO)+ ν (SiC) of R-(CH2)2-
Coupling product: chemical structure and observed 1H and 13C-NMR signals of the alkoxysilane fast blue RR coupling product

NMR, nuclear magnetic resonance; DMSO, dimethyl sulphoxide
Coating of thin layer chromatography plates with functionalised siloxanes
The coating of silica thin layer chromatography (TLC) plates was carried out as follows. Squares were cut out with a size of 2 × 2 cm, sprinkled with 100 μL of the substrate, dried for one hour at room temperature and were then placed for 24 h at approximately 105℃ in the oven.
Biotransformation of the immobilized substrates using MPO
The TLC plates with immobilized substrate were incubated in 100 μL potassium phosphate buffer (100 mmol/L; pH 7.1), 1.5 μL hydrogen peroxide solution (1% [v/v]) and 50 μL MPO (150 U/mL) or 150 μL buffer and 1.5 μL of hydrogen peroxide (1% [v/v]) for the blank. Additionally, oxidation of immobilized dyes was carried out with infected and non-infected WF samples, using 100 μL potassium phosphate buffer (100 mmol/L; pH 7.1) and 50 μL WF. The conversion of immobilized substrates was quantified with a spectrophotometer (ColorLite sph850, Katlenburg-Lindau, Germany) based on the colour difference between the reference and the sample, giving ΔE values according to the CIELab concept. Visual inspection of the samples after one day of incubation at room temperature was carried out. The experiment was repeated three times.
In situ generation of hydrogen peroxide with cellobiose dehydrogenase
Cellobiose dehydrogenase (CDH) (EC 1.1.99.15) was used for generation of hydrogen peroxide required by MPO. H2O2 production was based on a modified method published by Pricelius et al. 26 Essentially, the reaction mixtures as described above contained cellobiose (5 mmol/L), desferrioxamine mesylate (1 mmol/L) and 1.5 U/mL of CDH instead of hydrogen peroxide.
Results
MPO activity in WF
In this study, significantly higher MPO activity was detected in infected wounds when compared with non-infected wounds based on the oxidation of guaiacol using commercial MPO as standard. Eight wounds (four postoperative and four chronic wounds) were classified as not infected by the attending medical doctors. Four postoperative and four chronic wounds were described as clinically infected. Additionally, fluids from 10 non-infected blisters served as negative controls. MPO activity in infected WF was significantly (P ≤ 0.0005) higher (92.2 ± 49.0 U/mL) than in non-infected wounds (1.9 ± 1.8) and blisters (0.19 ± 0.5834 U/mL, P ≤ 0.0005), which was in good agreement with the clinical description. There was no significant difference between blisters and non-infected postoperative or chronic wounds (P = 0.021) regarding MPO activity levels. Similarly, there were no significant differences of MPO activity levels between infected chronic wounds and infected postoperative wounds (P = 0.89).
Novel MPO substrates for the detection of infection
Guaiacol is widely used for spectrophotometric quantification of MPO activity in general and was successfully applied for the detection of MPO activity in WF. In this study, commercial MPO as well as WF from infected and non-infected wounds were used for developing a reliable and yet fast diagnostic system for wound infection. Therefore, different and, in part, novel (for MPO) substrates like ABTS (10 mmol/L, measured at 410 nm), TMB (25 mmol/L, measured at 650 nm), LCV (1 mmol/L, measured at 590 nm) and fast blue RR salt (0.5 mmol/L, measured at 305 nm) were incubated with infected and non-infected WF samples. Fluids of five blisters served as negative controls (Figure 2). Additionally, haemoglobin content of the WF samples was determined and MPO activity corrected for the contribution of haemoglobin to substrate oxidation.
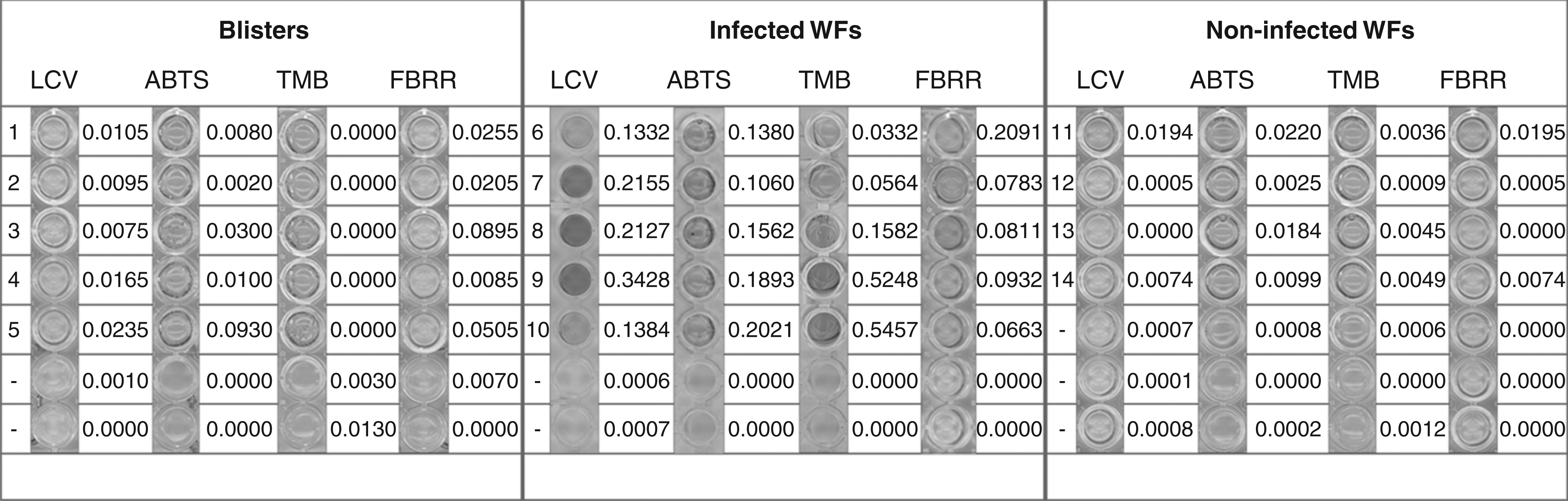
MPO activity in wound fluids (WFs) after incubation for 30 min shown as absorbance changes at specific wavelengths (ABTS [10 mmol/L, 410 nm], TMB [25 mmol/L, measured at 650 nm], LCV [1 mmol/L, measured at 590 nm] and FBRR [0.5 mmol/L, measured at 305 nm]). Samples were grouped as follows: blisters (1–5), infected WF samples (6–10), non-infected WF samples (11–14) and controls (−) containing substrate only. Contribution of haemoglobin to substrate conversion was subtracted. MPO, myeloperoxidase; ABTS, 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid); TMB, 3,3′,5,5′-tetramethyl-benzidine; LCV, leuco crystal violet; FBRR, fast blue RR
Using TMB and ABTS as MPO substrates, only in the case of infection was a clear colour change observed after an incubation time of 30 min. The coefficient of variation was 2.4% (ABTS) and 0.69% (TMB) in infected and 2.15% (ABTS) and 1.24% (TMB) in non-infected wounds. These results accord very well with the clinical description and with the MPO activity determined with guaiacol as a substrate. TMB was more sensitive than ABTS. The substrate solution containing hydrogen peroxide was stable for at least 12 weeks at 4℃.
Incubation of infected WF samples with fast blue RR resulted in a clear pink coloration of the sample after 30 min of incubation, while there was only a very light coloration in the two other groups. The experiments were repeated five times with a coefficient of variation of 3.7% in infected and 7.2% in non-infected wounds. It is thus shown for the first time that fast blue RR can serve as a suitable and sensitive substrate for determining MPO activity in WF.
Using LCV as the MPO substrate, incubation with the WF samples resulted in the formation of crystal violet with a dark violet colour after 30 min of incubation. Non-infected WF samples and blisters showed only a very light coloration. Again, infected WF samples can clearly be distinguished from blisters and non-infected WF based on elevated MPO activity. The experiments were repeated five times, with a coefficient of variation of 5.28% in infected and of 6.7% in non-infected wounds.
Further oxidation of crystal violet with MPO resulted in an absorbance shift from 580 to 560 nm after one hour of incubation (150 U/mL) at 25℃ (Figure 3a).
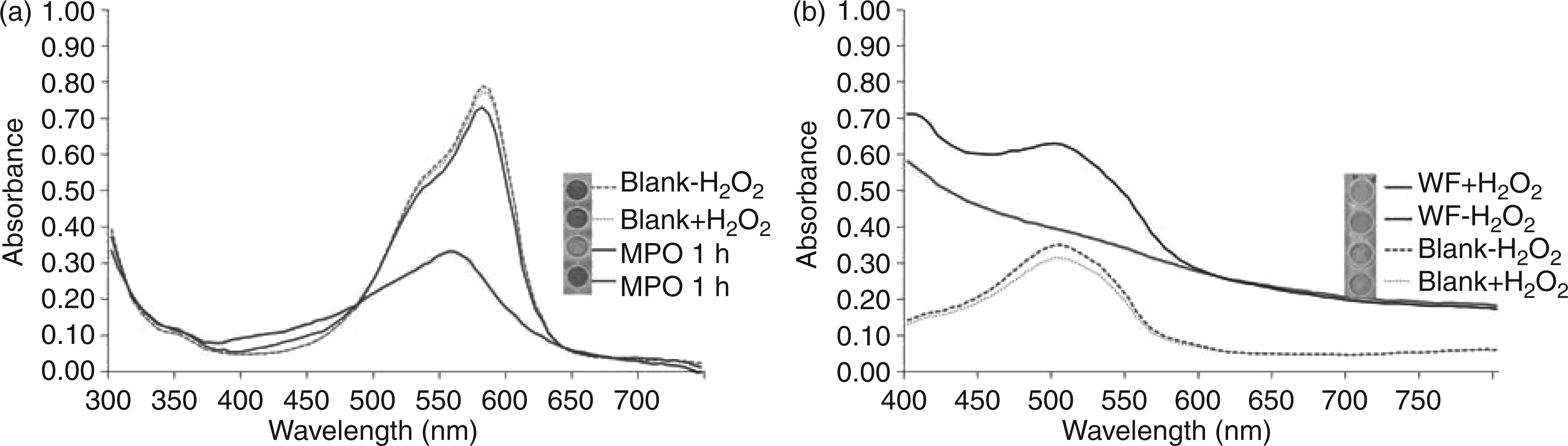
(a) Oxidation of crystal violet by myeloperoxidase (MPO) (150 U/mL) and colour change (inset picture) after incubation for 0 h (MPO 0 h) and one hour (MPO 1 h). (b) Oxidation of 2,7-dihydroxy-1-(4-hydroxyphenylazo)naphtalene-3,6-disulphonic and colour change (inset picture) after incubation with wound fluid (WF) in the presence and absence of hydrogen peroxide
This wavelength shift resulted in discoloration of the sample. H2O2 and buffer did not show any bleaching of the violet colour of crystal violet. Incubation with wound samples for 1 h did not lead to a significant colour change which is likely due to the lower MPO activity observed in WF, as longer incubation times indeed resulted in a clear colour change. Owing to the fact that there are no data for crystal violet as the MPO substrate, the catalysed oxidation of crystal violet by MPO was investigated by using HPLC-MS. The characteristic peak for crystal violet 372.53 (m/z) completely disappeared after incubation with MPO for 24 h. Incubation for six hours resulted in the appearance of a small peak at mass/charge ratio of 332.4, indicating a demethylated and deaminated molecule. After 24 h of incubation, this was no longer evident, suggesting a further degradation. The complete MPO-initiated break down of the molecule thus involved successive cleavage of all methyl groups followed by deamination (Figure 4). Finally, the backbone was cleaved.
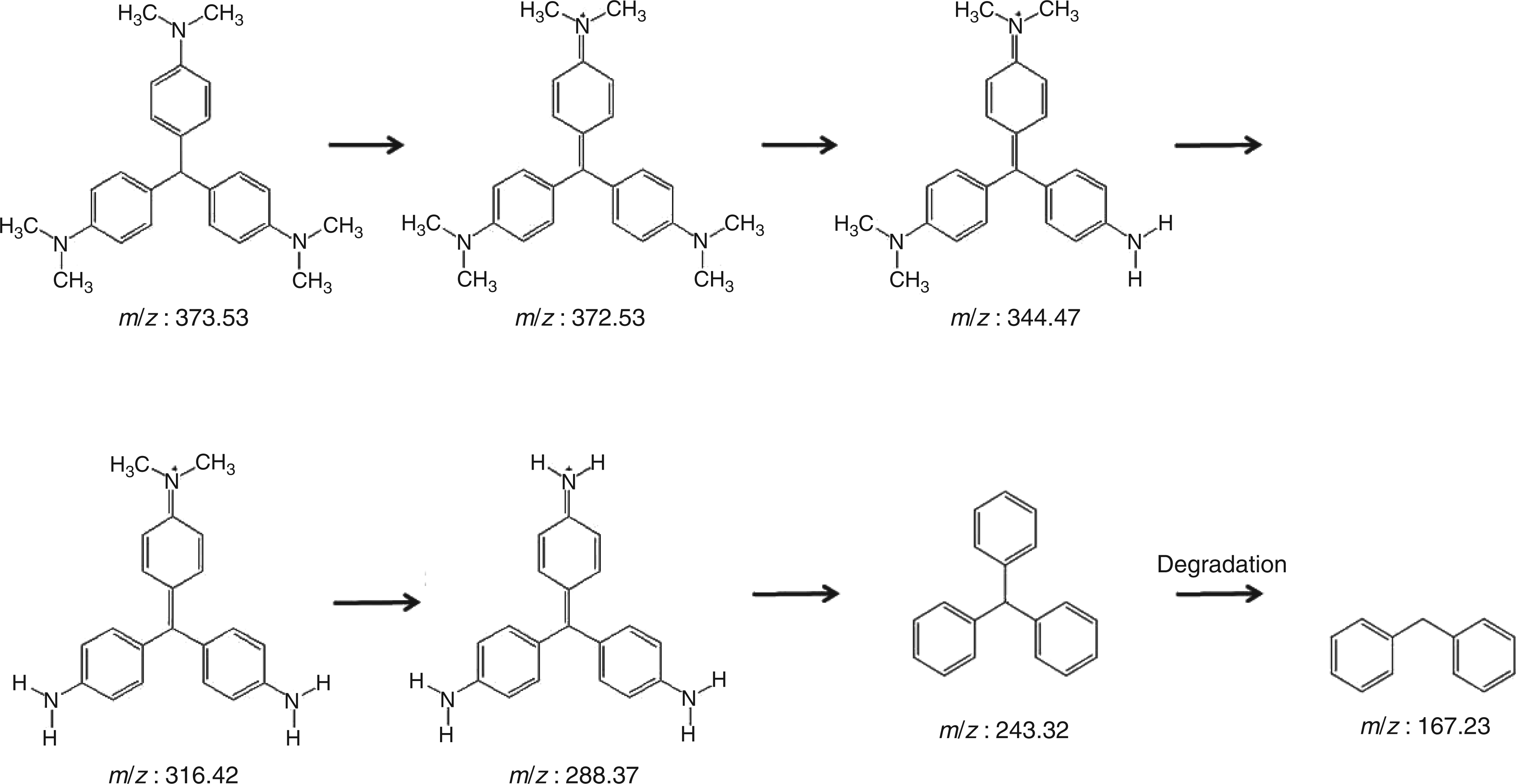
Hypothetical reaction pathway of myeloperoxidase (MPO)-catalysed oxidation of leuco crystal violet (LCV) (372.53 (m/z)) as determined with mass spectrometry
The reaction mechanism of MPO was studied further by means of oxidation of systematically substituted model substrates based on 2,7-dihydroxy-1-(4-hydroxyphenylazo)naphtalene-3,6-disulphonic acid. Among them, those derivatives carrying hydroxyl groups in ortho and para positions to the azo bond showed highest conversion after incubation with WF sample for 30 min (i.e. decolorization) as can be seen in Figure 3b.
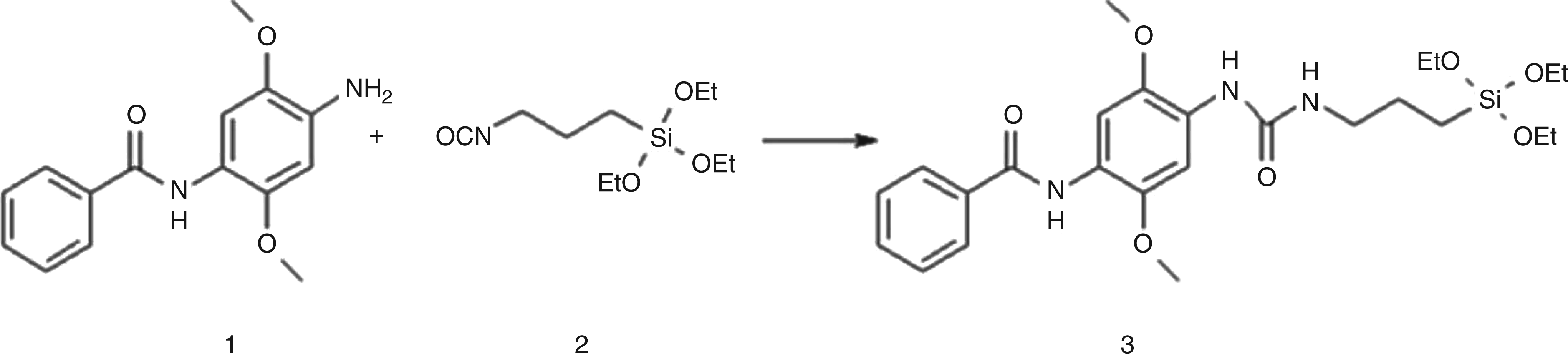
Fast blue RR (1) coupled to 3-(triethoxysilyl)propyl isocyanate (2) resulting in the stable urea product (3)
On the other hand, the meta-substituted analog, the methyl derivatives and the un-substituted derivative only showed marginal conversion.
On chromatographic and mass spectral analysis, all reactive substrates showed similar fragmentation patterns indicating lower-molecular-weight compounds after cleavage of the chromophoric bond. In all these cases, the 3,4,6-trihydroxynaphtalin-2,7-disulphonic acid (m/z 335; Figure 1b) was found at a retention time of 8.5 min. This compound, as the common structural feature among others, gave two major mass fragments with m/z 318 (dehydroxylated fragment) and m/z 188. Further fragmentation of this latter compound in a secondary mass spectrum led to a predominantly present fragment identified as 2, 3-dioxobenzene sulphonic acid (Figure 1c).
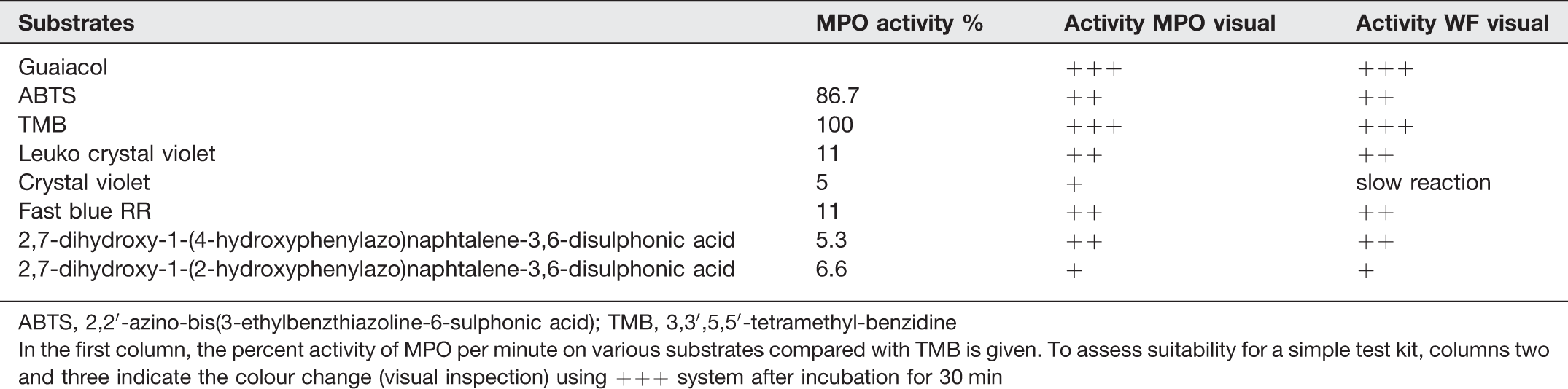
Comparing the studied MPO substrates regarding their potential in the assessment of WF enzyme concentrations, fast blue RR, 2,7-dihydroxy-1-(4-hydroxyphenylazo)-naphtalene-3,6-disulphonic acid and LCV seemed to be the most suitable (Table 2).
Comparison of various myeloperoxidase (MPO) substrates incubated with commercial MPO (10 U mL−1) or wound fluid (WF)
ABTS, 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid); TMB, 3,3′,5,5′-tetramethyl-benzidine
In the first column, the percent activity of MPO per minute on various substrates compared with TMB is given. To assess suitability for a simple test kit, columns two and three indicate the colour change (visual inspection) using +++ system after incubation for 30 min
Covalent immobilization of MPO substrates
Since immobilization of MPO substrates on solid materials would facilitate application of the diagnostic system (e.g. test strips, integration in bandages), coupling of the tested MPO substrates to carriers (i.e. siloxanes) was assessed. However, coupling of all investigated substrates, except fast blue RR, failed, since no stable products were obtained and/or they did not react with MPO (data not shown). Fast blue RR was successfully coupled to alkoxysilane isocyanates, resulting in a stable urea compound as indicated by 1H and 13C NMR analysis (Figure 5). The resulting functionalized siloxanes were polymerized on silica gel plates and glass slides as carriers. MPO was indeed able to oxidize the immobilized substrate after one day of incubation, resulting in a colour change towards dark red. This colour change corresponded to a ΔE of 17.95 (Figure 6). Additionally, incubation with infected and non-infected WF samples was performed. Again, a colour change was determined for infected WF samples, while there was no colour change in non-infected wound samples (data not shown).

Fast blue RR (N-(4-amino-2,5-dimethoxyphenyl) benzamide) coupled to alkoxysilanes was immobilized on silica gel. The conversion of immobilized substrates after incubation for one day was quantified with a spectrophotometer
Enzymatic in situ production of hydrogen peroxide
To improve storage stability of a diagnostic kit, a system for enzymatic in situ production of hydrogen peroxide was established. LCV was incubated either with commercial MPO or WF in the presence of CDH and cellobiose instead of hydrogen peroxide. Replacement of hydrogen peroxide by CDH gave an absorbance value of 0.708, while the usage of hydrogen peroxide in the buffer resulted in an absorbance of 0.56 after 60 min of incubation with MPO. Likewise, incubation of WF with MPO in the presence of CDH/cellobiose and hydrogen peroxide gave comparable absorbance changes.
Discussion
Wound infection is one of the most common reasons for the non-healing of a wound, leading to death of the patient in the worst case. It is widely assumed that WF has the potential to provide information which can be used as a diagnostic indicator for the overall status of a wound or as prognostic indicators.17–19
A significant difference in MPO activity between infected wounds and non-infected wounds or blisters was found in this study, accordingly well with the clinical description of the attending medical doctors. There was no significant difference between blisters and non-infected postoperative or chronic wounds regarding MPO concentrations. This observed difference between infected and non-infected wounds probably reflects the number of monocytes/macrophages and neutrophils in the tissue. As neutrophils are normally the first cells at the site of inflammation or injury, 6 the infiltration of neutrophils to injured tissue is characteristic of the host defence and inflammation. 7 Neutrophils and monocytes are predominant cell markers of wound infection. 28
Therefore, elevated concentrations of MPO released by activated neutrophils are an indicator of the inflammatory process.3,29
Currently, the detection of wound infection is based on evaluation of the well-known signs of inflammation like rubor (redness), calor (heat), tumour (swelling) and dolor (pain) by medical doctors and/or time-consuming procedures requiring special machinery like identification of the causative organism(s) after wound swabbing (Collier) or determination of serum CRP.20,21 However, both methods are not suitable for rapid diagnosis of wound infection. 22
Therefore, we decided to use MPO as a potential marker of the inflammatory process, in order to develop a fast diagnostic tool indicating the presence of neutrophils before other manifestations of infection. Consequently, screening for new suitable substrates for MPO detection in WF was carried out.
Guaiacol is a well-known substrate suitable for spectrophotometric measurement of MPO activity in WF. However, apart from its toxicity, we and others have experienced that the formed colour is not stable in some cases. It is well known that peroxidise-catalysed oxidation of guaiacol proceeds via coloured oligomers like tetraguaiacol30,31 and polymers that can precipitate. 32 Therefore, ABTS and TMB were tested for their ability to serve as MPO substrates in a fast diagnostic tool. Benzidine and its derivatives are widely used for both detection and quantification of peroxidase activity. However, most have carcinogenic and mutagenic properties, presenting serious drawbacks to their use in the laboratory. 33 One of the derivatives, TMB was chosen, since it is known to be neither carcinogenic nor mutagenic. 34 In addition, a high sensitivity for measuring the chlorination- and peroxidase activity of MPO compared with benzidine-based assays has been reported. 35 Comparing the sensitivity of ABTS and TMB with respect to reaction times and absorbance changes after incubation with WF, the latter seems to be more suitable for a diagnostic system.
In this study, crystal violet and its leuco form were for the first time shown to be converted by MPO. In agreement with previous results for horseradish peroxidase, 36 N-demethylation seems to be the first step in crystal violet degradation. This is supported by the fact that the absorption maximum of the dye underwent a hypsochromic shift since N-demethylation products are known to have absorption maxima at wavelengths lower than that of crystal violet. 37 Thereafter, step-wise de-amination finally leads to the breakdown of the backbone as indicated by LC-MS data. Oxidation of the leuco form was found to be a considerably faster process which is in agreement with data reported for other peroxidases. 38 Consequently, regarding assessment of wound infection, LCV was found to be more suitable, giving clearly visible colour changes when incubated with infected WF for only 10 min.
Most of the substrates used in this study can also be oxidized by other peroxidases like horseradish peroxidase, laccase or eosinophil peroxidase. However, eosinophils have functional roles in host defence against helminth infections and in the immunpathogenesis of allergic diseases. Other peroxidases do not have major roles in host defence against bacteria 39 and therefore do not interfere with assays for MPO in WF.
In addition, an influence of haemoglobin potentially contributing to oxidation of MPO substrates was shown for most of the substrates except guaiacol. As some of the tested WF samples contained varying amounts of haemoglobin, the influence of haemoglobin on the substrates was determined and subtracted from the overall activity to calculate contribution made by MPO. Nevertheless, a clear distinction between infected and non-infected diluted wound samples was possible.
In an attempt to obtain more information on the structural requirements for substrates to be oxidized efficiently by MPO, model substrates were synthesized. Based on 2,7-dihydroxy-1-phenylazonaphtalene-3,6-disulphonic acid, systematically substituted derivatives were constructed in order to gain mechanistic information on MPO-catalysed oxidation. In agreement with our previous investigations involving laccases, 40 the nature and position of the substitution on the molecule had a dramatic influence on MPO-catalysed oxidation. Hydroxyl groups and, to a lower extent, methyl-substituted derivatives, were readily oxidized while there was no detectable reaction with derivatives carrying chlorine or nitro groups. This is in agreement with results previously found for laccases. 27 Most likely, electron donating substituents can stabilize the aromatic cation formed after two subsequent electron abstractions. 41
Hydrogen peroxide is a required co-substrate in MPO-catalysed oxidation of all the above substrates. To improve storage stability of a diagnostic kit, enzymatic in situ production of hydrogen peroxide was investigated. A recently reported CDH (EC 1.1.99.15) from Myriococcum thermophilum which accepts a wide range of sugars and polysaccharides was used to produce hydrogen peroxide. 26 Indeed, it was possible to replace hydrogen peroxide in the assay solution by addition of this enzyme together with cellobiose.
Immobilization of MPO substrates would have advantages both in terms of simpler handling (e.g. test strips) as well as when the diagnostic device is to be integrated in bandages. In the latter case, it will be essential that there is no dye release into wounds. Here we showed for the first time that fast blue RR can be covalently coupled to alkoxysilanes and still remain a substrate for MPO. This functionalized alkoxysilane can easily be polymerised onto a large variety of materials ranging from polyesters and cotton as materials of bandages to glass or silica gel as potential carriers in test strips.
Summarizing our results, we have demonstrated the potential of novel, partly immobilized MPO substrates for the detection of MPO activity in WF. Using these substrates, visible colour changes indicate elevated enzyme concentrations in infected wounds after very short incubation times. These results can be the basis for the development of a diagnostic kit for early detection of wound infection.