
Abstract
Autosomal dominant hypercholesterolaemia (ADH) is due to defects in the LDL receptor gene (LDLR), in the apolipoprotein B-100 gene (APOB) or in the proprotein convertase subtilisin/kexin type 9 gene (PCSK9). The aim of this study was to identify and to characterize mutations at the origin of ADH in two Tunisian families. We found three genomic variations: (1) c.1845 + 1G > A, a splice site mutation in the LDLR gene and (2) two variations in the PCSK9 gene (p.Phe515Leu and p.Gly670Glu) that were both reported to be associated with high LDL-C levels. These results enlarge the spectrum of ADH-causative LDLR and PCSK9 variations in Tunisia. Our observations indicate that missense variations in the PCSK9 gene do not influence the clinical phenotype of ADH patients carrying a mutation in the LDLR gene.
Introduction
Autosomal dominant hypercholesterolaemia (ADH) (OMIM #143890) is characterized by raised serum LDL-cholesterol levels, and accelerated atherosclerosis that increased the risk of premature coronary heart disease (CHD). 1
The most prevalent underlying molecular defect of ADH consists of mutations in the LDL receptor gene, but mutations in the LDL receptor binding domain of the apolipoprotein B (apoB) (familial defective apolipoprotein B) 2 and PCSK9 have also been associated with familial hypercholesterolaemia (FH). 3 Other still unknown genes could also be involved. 4
The frequency of heterozygous FH in most populations is about 1/500, homozygous FH is rare (1 per millon), 5 but in Tunisia, a higher frequency has been reported (1/165) because of the high levels of consanguinity in this population. 6 Eight mutations causing FH in the LDLR gene have been reported in the Tunisian population. 7,8
In this study, we report two apparently unrelated families with severe hypercholesterolaemia. (We identified in probands three variations: one splice site mutation in the LDLR gene, and two missense variations in the PCSK9 gene.)
Materials and methods
Patients
Family members, who had given informed consent for the study, underwent clinical examination and plasma lipids levels were measured by enzymatic methods as previously described. 6
Based on these criteria, ADH was identified in two apparently unrelated families A and B, including nine and five individuals, respectively (Figure 1).
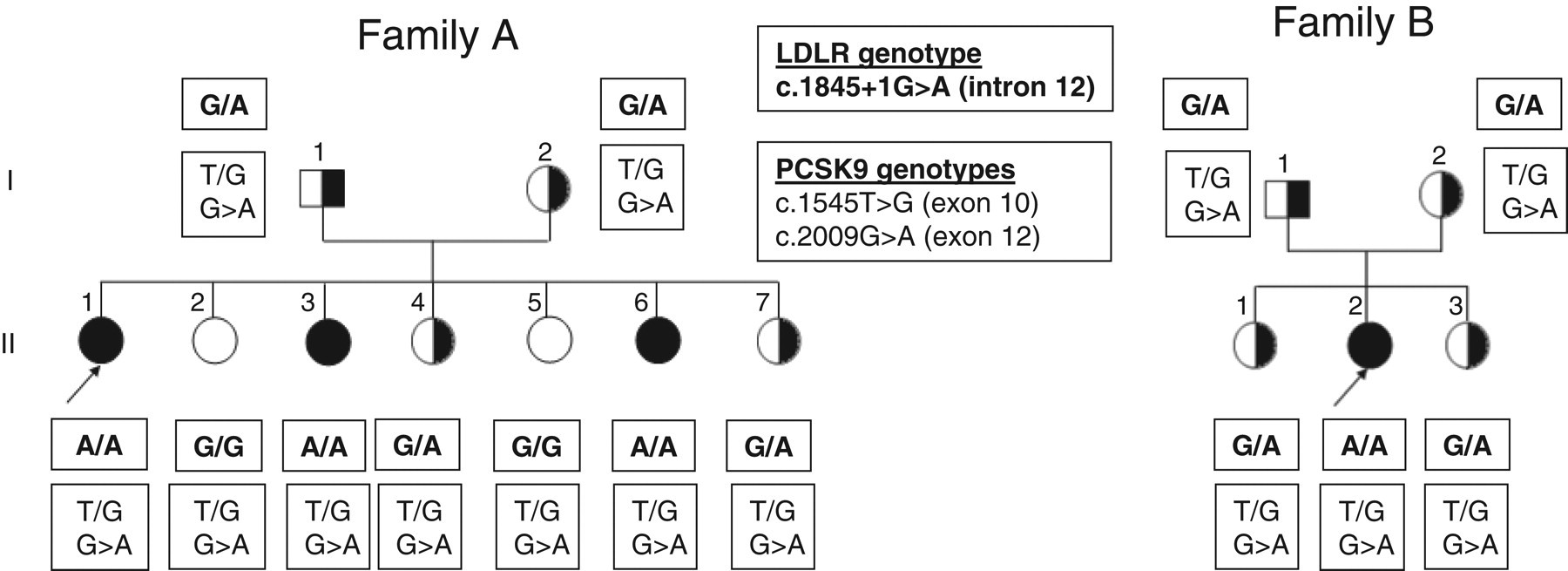
Filled and half-filled symbols represent clinically defined homozygotes and heterozygotes, respectively. Unblackened symbols indicate normocholesterolaemic individuals and probands are indicated by arrows
Sequencing for mutations
Genomic DNA was isolated from EDTA blood samples. Amplification of promoter and exons of the LDLR and the PCSK9 genes was performed. Automated sequencing of purified polymerase chain reaction (PCR) products (Sephadex G50 spin columns, Boehringer Manheim, Germany) was performed on sense and antisense strands according to the supplier's instructions (Applied Biosystems, France). The common mutation p.ArgR3527Gln in the APOB gene was screened using the restriction method.
Analysis of LDL receptor activity in cultured skin fibroblasts
Analysis of LDL receptor activity was performed on cultured skin fibroblasts obtained after skin biopsy from probands and a healthy donor. Cells culture, binding, uptake and degradation of 125I-LDL were determined.
Detection of the 1845 + 1G > A mutation
For rapid screening of this mutation, a PCR assay was designed using the Bgl II restriction enzyme (Biolabs, New England), because the c.1845 + 1G > A mutation generates a new restriction site for Bgl II. The amplification was performed using a forward primer: 5′ GCACGTGACCTCTCCTTATCCACTT 3′ and a reverse primer 5′ CGATCTCGTACGTAAGCCAGATCTC 3′ with a mismatch substitution of C to G.
The reaction mixture for the PCR was made with 1 μg of genomic DNA; 2.5 U Taq polymerase (Promega, France) and 0.2 mmol/L of each dNTP. The PCR conditions were 35 cycles: 95°C/30 s, 55°C/30 s, 72°C/30 s.
The amplified product was treated with 10 U Bgl II at 37°C overnight, and obtained fragments were run under 120 V on 10% polyacrylamide gel, and then stained with silver nitrate.
Haplotype analysis
Haplotypes were constructed by segregation of biallelic polymorphisms of the LDLR gene.
Results
In the nine patients from family A and five patients from family B, the p.Arg3527Gln mutation in the APOB gene was not found in any subject. Direct sequencing of the LDLR gene showed only one variation in intron 12. This mutation is a single base substitution G to A in the first base of the 5′ splice donor site of intron 12 (c.1845 + 1G > A). This mutation has been reported by Hobbs (FH-Tunis), and associated with abnormal mRNA.
Using a mismatch primer, this G > A variant creates a new restriction site for the endonuclease Bgl II, and the PCR product (907 bp) is restricted to three fragments (748, 140 and 19 bp) in the mutant allele, whereas in the normal allele it is restricted to two fragments (748 and 159 bp). The c.1845 + 1G > A substitution was co-segregated with the clinical and biological status of ADH.
Direct sequencing of the PCSK9 gene showed that all patients carried two alleles: the c.1545T > G transition in exon 10, causing a phenylalanine for leucine conversion at codon 515 (p.Phe515Leu) and the c.2009G > A transition in exon 12, causing a glycine for glutamic acid conversion at codon 670 (p.Gly670Glu). Both mutations were associated with high LDL cholesterol levels.
Analysis of seven RFLPs at the LDLR locus showed that the same haplotype co-segregated with the c.1845 + 1G > A mutation in the two families, thus providing evidence for a founder effect.
Incubations of the cultured fibroblasts from patients II-1 (family A) and control subject with 125 I-labelled LDL showed that binding, uptake and degradation of LDL were extensively diminished for the FH subjects. At 4°C, binding of iodinated LDL on fibroblasts of probands II-1 from family A was less than 2% of the activity found in normal cells. Skin xanthoma was present in the four homozygotes, and CHD was present in 75% of them. None of the heterozygotes had CHD and only two had xanthomas.
Plasma lipids levels were expressed as multiple of median (MoM) for age and gender of a reference population. 5 As expected, the total cholesterol mean MoM for homozygotes, 4.0 ± 0.5 (n = 4), was more than twice the mean MoM for heterozygotes, 1.5 ± 0.3 (n = 8). For LDL-C mean MoM for homozygotes, 6.0 ± 1.1, was almost three times the mean MoM for heterozygotes, 1.9 ± 0.5. The mean MoM for HDL-C and triglycerides levels were near 1 for both homozygotes and heterozygotes.
We also compared mean MoM of lipid concentrations of patients with combined variations in the LDLR and PCSK9 genes with mean MoM of lipid concentrations of Tunisian FH patients with known mutation in the LDLR gene. 9 We noted that for combined heterozygote (N = 8) mean MoM of total cholesterol were similar to heterozygote Tunisian FH patients (N = 42) (1.5 ± 0.3). Concerning LDL-C level, the mean MoM for combined heterozygote (1.9 ± 0.5) was slightly higher than that for heterozygote FH patients (1.8 ± 0.4). The same observation was noted concerning HDL-C (0.9 ± 0.1 for combined heterozygote versus 0.8 ± 0.3 for heterozygote Tunisian FH patients). The mean MoM of triglycerides was slightly higher in heterozygote Tunisian FH patients than in combined heterozygote (1.0 ± 0.7 versus 0.6 ± 0.2).
Concerning homozygous, the analysis of these levels showed that, the mean MoM for total cholesterol and LDL cholesterol were similar for combined homozygote LDLR/heterozygote PCSK9 (N = 4) and homozygous FH Tunisian patients (N = 26). Mean MoM for triglycerides levels was lower for combined homozygote LDLR/heterozygote PCSK9 than for homozygous FH Tunisian patients (1.0 ± 0.5 versus 1.6 ± 0.8). The mean MoM for HDL cholesterol was higher for combined homozygote LDLR/heterozygote PCSK9 than for homozygous Tunisian FH patients (0.8 ± 0.3 versus 0.6 ± 0.3).
Discussion
In this article, we describe the genotyping of two apparently unrelated families from south Tunisia, with a clinical phenotype and family history, suggesting the diagnosis of ADH.
In the LDLR gene we identified, in the two probands, a splice site mutation: c.1845 + 1G > A in the first base of the 5′ donor site of intron 12. This base substitution changes the highly conserved dinucleotides GT to AT. The mutation results in aberrant splicing of the mRNA (size 7.5 kb). As previously reported, 10 we confirmed that LDL receptor activity in cultured fibroblast in homozygote carrier (family A, II-1) was less than 2%. This mutation has also been reported for a New Zealand subject from European origin. 11
Exon 12 is located in the second domain of the LDL receptor, which contains the region homologous to the EGF precursor. A total deletion of this domain does not affect the transport or maturation of the receptor, but the acid-dependent association of the lipoprotein in the endosomes and recycling of the receptor are altered. This domain is also required to keep the proper conformation needed for the apoB binding.
PCSK9, a serine protease, has recently gained attention because of its major role in the regulation of plasma LDL cholesterol level and causation of CHD. The link between PCSK9 and plasma LDL-C levels was first established by the discovery of missense mutations in the PCSK9 gene that were present in patients with ADH. 3 These mutations were speculated to result in a gain of the function of PCSK9. Subsequently, loss of function mutations in the PCSK9 gene were discovered that are associated with low plasma LDL-C levels and significantly reduced CHD. 12
In this study, two variations in the PCSK9 gene have been identified. Both PCSK9 variations found in the patients are located in the c-terminal domain of PCSK9 and involve amino acid residues, which are highly conserved in humans, mice and rats.
The p.Gly670Glu variation is a frequent polymorphism, and it was reported to be associated with high levels of cholesterol, especially in men, in a German population. 13 Conversely, a recent study in a Chinese population showed that this same polymorphism was associated with low LDL-C levels and is not a risk factor for coronary artery disease in ethnic Chinese in Taiwan. 14
The p.Phe515Leu is a rare variant and was found in only one hypercholesterolaemic African-American subjects. 15
Additive effects of mutations in the LDLR and PCSK9 genes on the phenotype of FH have also been described and are associated with a severe phenotype similar to homozygous FH. In this report, probands in two families from south Tunisia carrying variations in the LDLR and PCSK9 genes presented with a severe phenotype of hypercholesterolaemia. We showed that phenotype of ADH is due especially to the splice mutation in the LDLR gene, and that variations in the PCSK9 gene do not influence the phenotype of ADH in these Tunisian patients.
DECLARATIONS