
Abstract
Superficial siderosis is an increasingly common diagnosis due to advances in imaging. Resulting from the presence of blood in the subarachnoid space, it leads to progressively debilitating ataxia, deafness and myelopathy. Magnetic resonance imaging has revolutionized diagnosis and management of this condition. In one-third of patients with this diagnosis, no subarachnoid bleed can be identified. A lack of sensitivity in our ability to detect subarachnoid bleed is a possible explanation in this group of patients. A novel approach would be to consider defects in the body's defense against intracranial iron overload as a possible cause of this disease. A disproportionately few patients develop superficial siderosis compared with the number who develop subarachnoid bleed from various clinical conditions. Normal physiological protective mechanisms exist in the central nervous system to protect it from damage by blood, haeme or free iron. Deficiency and defect of these protective mechanisms can provide insight into the pathogenetic mechanism of the group of patients where no subarachnoid haemorrhage is identified.
Introduction
Superficial siderosis is caused by prolonged or recurrent low-grade bleeding into the cerebrospinal fluid (CSF). Underlying causes include recent or previously resected central nervous system (CNS) tumours, CSF cavitary lesions (meningocoele, pseudomeningocoele), vascular malformations, CNS trauma, subarachnoid haemorrhage, intracranial or spinal surgery and brachial plexus or nerve root injury. The source of bleeding remains unknown in one-third of patients. 1 Haemosiderin arising from the bleed causes oxidative cellular damage leading to symptoms. Despite the central role of haemosiderin, superficial siderosis remains a distinct disease from haemochromatosis.
Deafness and ataxia are ascribed to damage of the VIII cranial nerve and the cerebellar tracts, and are present in about 80% of patients. Myelopathy which may lead to lower motor neuron weakness or upper motor neuron spasticity is seen in half. Less common symptoms include bladder incontinence or retention, headache, anosmia, bowel dysfunction, ageusia, other cranial nerve abnormalities, cerebellar dysarthria, nystagmus and a distinct form of cognitive defect involving speech, executive function and visual recall. 2 The prominent injury of the eighth cranial nerve is due to its long course through CSF within the pontine cistern and its investment by numerous microglial cells that become haemosiderin-laden. The olfactory nerve suffers similar damage. The optic nerve is spared because it is not exposed to CSF.
Case
A 64-year-old male patient presented with progressive difficulty in walking and decreased hearing, developing over two years. He had down-beating nystagmus, bilateral sensorineural deafness and hyperactive lower limb reflexes. The gait was markedly ataxic with tendency to fall. Finger–nose and heel–shin tests were abnormal bilaterally. Cognitive examination was normal.
He had adequately treated hypothyroidism, no history of CNS trauma or surgery and was not on phenytoin. There was no history of malignancy. He did not drink alcohol, and had no family history of ataxia.
Routine blood counts and serum biochemistries were normal. Thyroid function tests, vitamin B12, folic acid and thiamine concentrations were normal. Laboratory tests for tertiary syphilis and antigliadin antibody syndrome were negative. 3 A T2-weighted magnetic resonance imaging (MRI) revealed a sharp rim of hypointensity over the surfaces of the cervical spinal cord, brainstem, cerebellar hemispheres and portions of the cerebral hemispheres, consistent with superficial siderosis of CNS (Figures 1 and 2). Serum iron studies were normal. The CSF was clear. There was no pink or yellow discolouration. On microscopy it was acellular. The biochemical examination revealed normal values for pH, protein, sugar and electrolytes. Audiometry revealed bilateral sensorineural hearing loss.
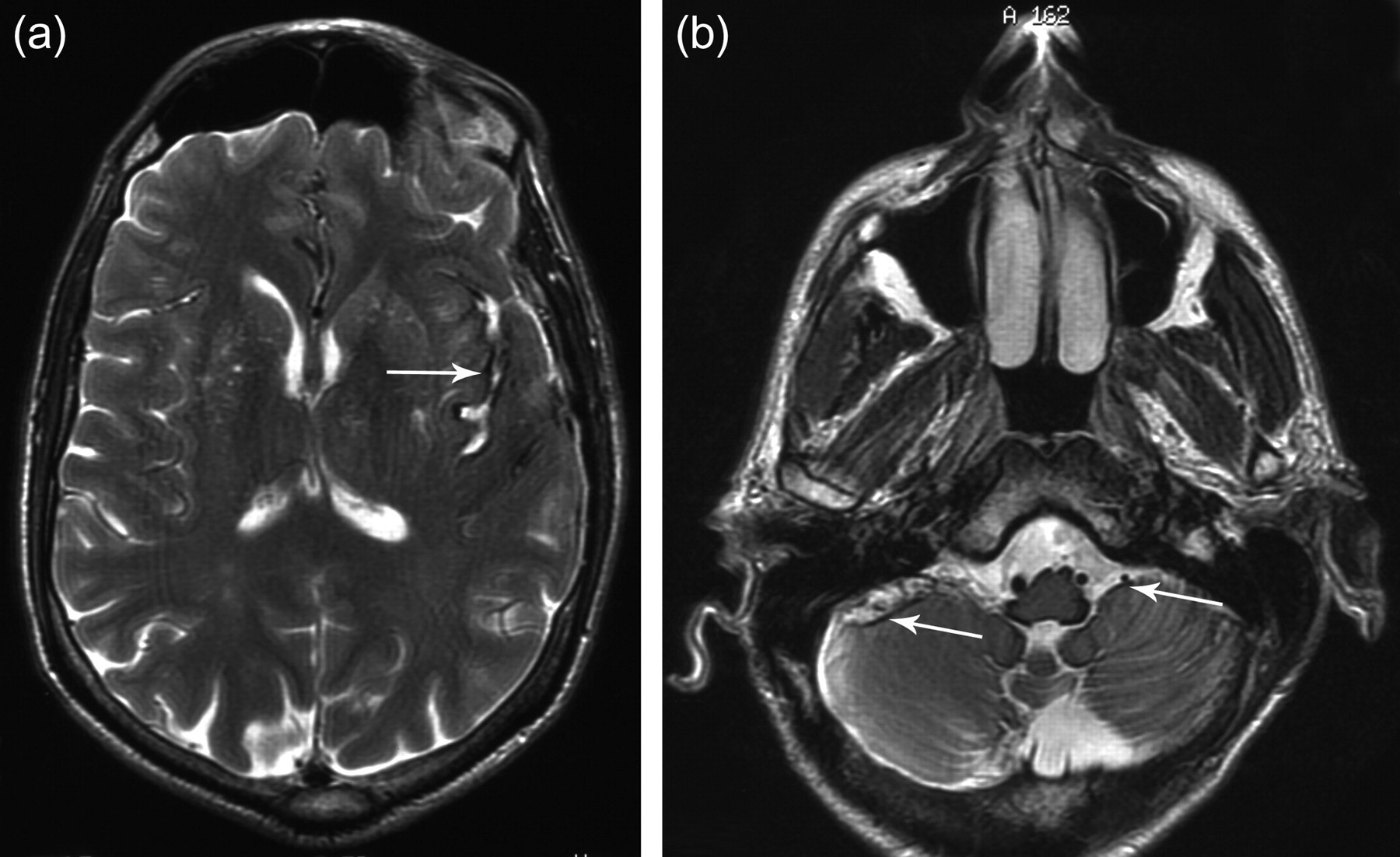
Magnetic resonance imaging T2-weighted images showing haemosiderin deposition (arrows) in axial view through the level of the insular cortex (a) and anterior margin of the cerebellum (b)
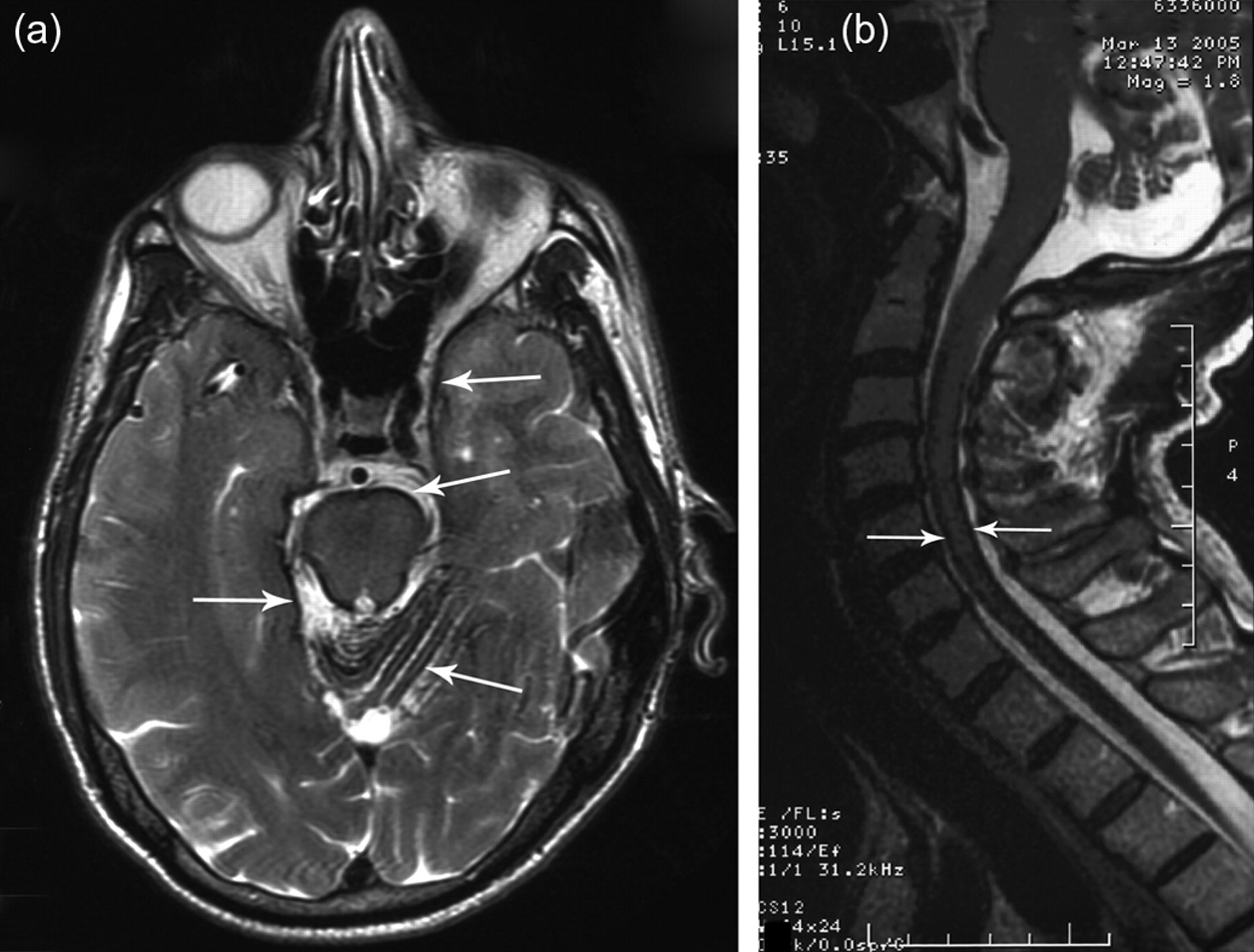
Magnetic resonance imaging T2-weighted images showing haemosiderin deposition (arrows) along cerebellar folia, brainstem, cerebral surfaces (a) and cervical spinal cord (b)
Subsequent CSF studies have remained normal and his disease has remained stable for the last one year. He uses a cane for mobilization, performs all activities of daily living and has retained bladder and bowel functions. The patient made an informed decision not to take trientine or undergo CSF shunting.
Discussion
Our patient had the classical presentation of slowly progressive cerebellar ataxia, sensorineural hearing impairment and myelopathy of the lower limbs. The clinical triad of ataxia, deafness and myelopathy is seen together in less than 50% patients. Clinical findings along with characteristic MRI findings confirmed the diagnosis of superficial siderosis. There was no hypogonadism, mental deficiency, piebaldism or pigmentary skin changes – signs seen in hereditary hearing loss with ataxias. 4
Prior to MRI technology, the diagnosis of siderosis was made at autopsy. A dark lining of the meningeal surfaces on T2 images is due to the paramagnetic nature of haemosiderin. MRI-FLAIR (fluid-attenuated inversion recovery) imaging suggests the diagnosis and should prompt MRI-GRE (gradient echo) sequence to confirm. 5,6 Increased use of neurosurgical procedures and improved survival after such procedures have led to a greater prevalence of superficial siderosis. However, MRI, MR angiography, computed tomography myelography and angiography may all fail to define a causative lesion, as in our patient.
Animal experiments have confirmed the role of recurrent bleed in the subarachnoid space as cause of superficial siderosis. 7 Cerebellar Bergmann cells, microglia and superficial astrocytes pick up subarachnoid blood. Intracellularly the haeme is broken down to free iron by the enzyme haeme oxygenase. Free iron causes toxic injury to the cell through production of reactive oxygen species. Both iron and haeme products cause neurodegenerative injury. 8 The free iron upregulates ferritin production – an evolutionary adaptation for cell preservation. Persistent free iron exposure leads to change in ferritin production from heavy subunit to light subunit. The light ferritin subunits allow long-term iron storage as haemosiderin deposits. While such storage provides short-term protection, in the long run this leads to damage. 9 Ferritin releases iron during oxidative stress further aggravating neuronal damage. 10
The MRI, early in the evaluation of a patient with ataxia and hearing loss, will quickly help to identify the presence of superficial siderosis. This practically eliminates the search for more esoteric causes of ataxia and associated progressive deafness. The work-up of such a patient should include an immediate comprehensive search for a source of CNS bleed. Quick identification and surgical repair of CNS bleeding sources are of paramount importance in preventing progression of symptoms. A CNS lesion may not be found in one-third of patients with superficial siderosis. Chronic subarachnoid haemorrhage can be intermittent, unassociated with detectable CNS lesions and the CSF may be entirely normal between episodes of intermittent, chronic subarachnoid haemorrhage. Repeat CSF examinations will be necessary to identify the patient who may have low-grade intermittent subarachnoid bleeding.
When no bleeding source can be identified the treatment becomes conjectural. Anecdotally proposed treatments include desferrioxamine, trientine and lumbar–CSF shunting. Tin protoporphyrin has been tried in the experimental rabbit model of superficial siderosis. Superficial siderosis is hypothesized to result from the toxic effects of blood in the subarachnoid space. CSF shunting will reduce prolonged exposure to blood and hence be theoretically helpful. The reported patient underwent lumboperitoneal shunt with a reservoir. CSF was analysed for cell counts, protein, glucose, bilirubin, ferritin and haemosiderin at admission and at follow-up. The patient was followed up with audiometry, visual-evoked potential and functional assessment. Significant change in CSF constituents was absent. There was some improvement in audiometry and functional assessments, visual responses being too inconsistent for analysis. The study was limited by single subject, late stage of disease, short follow-up and lack of standardized measurable outcomes. 11
Success of chelating agents is also rare. A patient of superficial siderosis presenting with ataxia, deafness, dysarthria and haemifacial spasm was treated with six months of trientine. 12 The clinical picture remained stable and haemifacial spasm disappeared. There was some reduction in CSF iron and ferritin concentrations. Whether CSF iron and ferritin can be used as parameters to follow the treatment of superficial siderosis remains controversial. 9 High CSF concentrations of iron and ferritin are also seen in malignant and inflammatory conditions of the CNS. 13
Superficial siderosis remains a rare disease. Since the original report in 1908, only 270 cases have been reported in 99 y between 1908 and 2006. 1 Considering the number of patients who have a CNS bleed from the various causative lesions of superficial siderosis, this remains a tiny fraction even if we account for a significant number of asymptomatic cases. There is no other hypothesis other than low-grade CNS bleeding as cause of this disease. However, there remains a great lacuna in our knowledge of physiological mechanisms that exist to protect the brain from iron-mediated injuries. An outline of the physiological mechanism of iron metabolism in the brain is shown in Figure 3. 14,15 Two possible hypotheses based on CNS bleed can account for the pathogenesis of one-third cases of superficial siderosis where we are unable to find a cause – (1) the CNS bleeding is too small to detect with available investigative modalities and (2) abnormalities in the physiological protective pathways that exist in the CNS to attenuate damage caused by blood, haeme or free iron. The iron metabolism and storage in different brain cells involves complex interaction between multiple factors. Available information on types of brain cells, forms of stored iron, receptor for uptake and extrusion are summarized in Table 1. 14,15 Numerous lacunae exist in our knowledge of iron metabolism in the brain. Defects in uptake, storage and extrusion of iron can all be pathogenic for superficial siderosis. This may provide insight regarding drugs that can affect various receptors and enable us to treat this disabling condition.
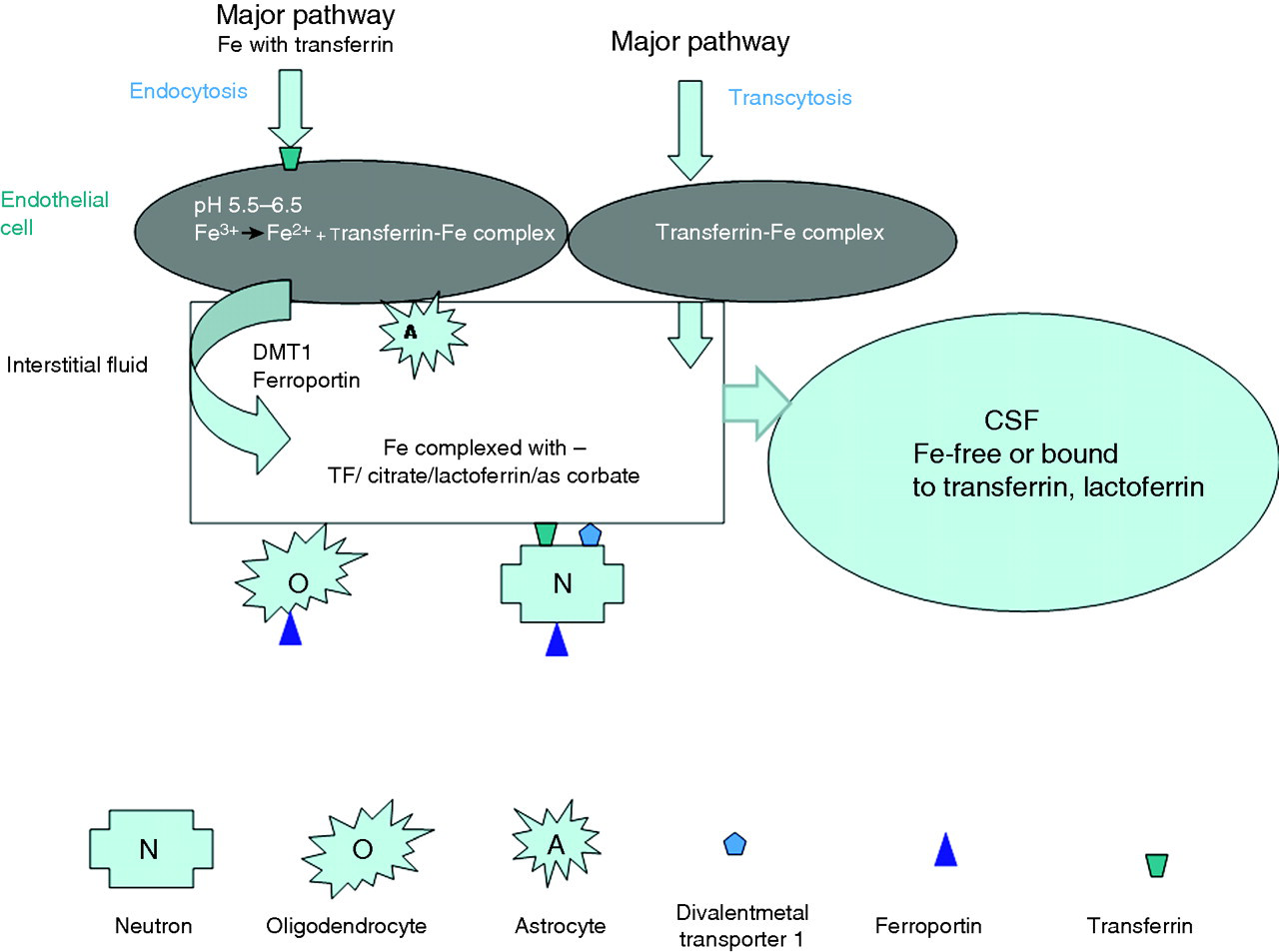
Iron is taken up by the endothelial cells (EC) at the blood–brain barrier by the transferrin (TF) receptors. This pathway forms the major pathway; transcytosis (also known as vesicular transport) is considered to be a minor pathway. In the EC, iron is released from TF in the presence of acidic pH. The apotransferrin (aT) is recycled and iron is released into the brain interstitial fluid at the abluminal surface of EC. The release at the abluminal surface can be mediated by receptors (divalent metal transporter 1 (DMT-1), ferroportin) or by astrocytes in the presence of citrate, acidic pH and ATP. In the interstitial fluid (IF) iron exists as complex with TF, lactoferrin, citrate and ascorbate. Between IF and brain cells, iron is cycled through receptors as shown. Iron is removed from the brain through CSF either in the free form or complexed with ferritin, lactoferrin. It can also be removed by microglial and other phagocytic cells
Iron metabolism – uptake, storage and removal from various brain cells
TF, transferrin; EC, endothelial cells; DMT-1, divalent metal transporter 1
Possible cause of excess iron storage and pathogenesis of superficial siderosis is proposed
DECLARATIONS