
Abstract
Central pontine myelinolysis (CPM) has been described in alcoholic patients and in the aftermath of rapid correction of chronic hyponatraemia. We describe a case of CPM occurring secondary to nephrogenic diabetes insipidus (DI), which developed as a consequence of severe hypokalaemia. A 63-year-old man with alcohol dependence was admitted to hospital with severe pulmonary sepsis and type 1 respiratory failure. On admission, he had euvolaemic hyponatraemia of 127 mmol/L, consistent with a syndrome of inappropriate antidiuretic hormone secondary to his pneumonia. Following admission, his plasma potassium dropped from 3.2 to a nadir of 2.3 mmol/L. Mineralocorticoid excess, ectopic adrenocorticotrophic hormone production and other causes of hypokalaemia were excluded. The hypokalaemia provoked significant hypotonic polyuria and a slow rise in plasma sodium to 161 mmol/L over several days. Plasma glucose, calcium and creatinine were normal. The polyuria did not respond to desmopressin, and subsequent correction of his polyuria and hypernatraemia after normalization of plasma potassium confirmed the diagnosis of nephrogenic DI due to hypokalaemia. The patient remained obtunded, and the clinical suspicion of osmotic demyelination was confirmed on magnetic resonance imaging. The patient remained comatose and passed away 10 days later. This is the first reported case of nephrogenic DI resulting in the development of CPM, despite a relatively slow rise in plasma sodium of less than 12 mmol/L/24 h. Coexisting alcohol abuse, hypoxaemia and hypokalaemia may have contributed significantly to the development of CPM in this patient.
Introduction
Central pontine myelinolysis (CPM) is a well described, potentially devastating consequence of rapid alterations in plasma osmolality/tonicity, classically occurring secondary to the excessively rapid correction of chronic hyponatraemia. 1 The cells of the central pontine region are particularly susceptible to fluctuations in osmolality, and the resultant clinical manifestations can vary from mild tremors, to progressive quadraparesis, to locked-in syndrome and to death in very severe cases. The potential for iatrogenic aetiology in the syndrome is reflected by the exponential rise in the reports of CPM that closely followed the widespread introduction of intravenous fluid therapy in the 1950s. 1 However, sodium replacement is not the sole cause of CPM, and a number of other co-morbid conditions have been identified to increase the individual's risk of developing the syndrome. These co-morbidities include alcohol abuse, malnutrition, liver transplantation, alterations in plasma glucose and hypokalaemia. 2–5
The only previously published case involving both diabetes insipidus (DI) and myelinolysis was reported by Chang et al. 6 They described the presentation of central DI secondary to lymphocytic hypophysitis in an otherwise well patient. The central DI in this case resulted in severe hypernatraemia and subsequent radiological changes that were consistent with extrapontine myelinolysis. Here we present the first reported case of CPM occurring secondary to nephrogenic DI caused by severe hypokalaemia, which is, in and of itself, an independent risk factor for CPM.
Case report
A 63-year-old man was admitted with a clinical and radiological diagnosis of severe pulmonary sepsis and type 1 respiratory failure. His past medical history was significant for alcohol dependency. On clinical examination he had coarse crepitations in his right lung base. He was clinically euvolaemic, and had no obvious signs of chronic liver disease. He was treated with oxygen, vitamin B complex (5-day intravenous regimen), and broad-spectrum intravenous antibiotics.
On admission he was noted to be hyponatraemic with the plasma sodium of 127 mmol/L (reference range 135–145) and a urea of 5.1 mmol/L (reference range 2.5–8.5). He was not on any medications noted to cause hyponatraemia. Given his euvolaemic status, his pneumonic process, his natriuresis and his subsequent endocrine testing, his low sodium was felt to be consistent with the syndrome of inappropriate antidiuretic hormone secondarily to his pneumonic process. Of note, this patient subsequently underwent computed tomography (CT) of the thorax and brain, magnetic resonance imaging (MRI) of the brain and postmortem examination, and no evidence of neoplastic lung disease or intracranial pathology as an alternative cause for his hyponatraemia was found. Additionally, there was no radiological evidence of cirrhosis on liver ultrasound. With an ongoing pneumonic process the patient received intravenous fluid therapy consisting of isotonic saline.
His initial plasma potassium was 3.2 mmol/L (reference range 3.2–5.3), which subsequently dropped to a nadir of 2.3 mmol/L within 24 h of admission, and severe hypokalaemia persisted in the days that followed despite intravenous replacement (Figure 1). His hypokalaemia was felt to stem from severe respiratory alkalosis, with a PH rising to 7.55 (secondary to tachypnoea from worsening respiratory status and type 1 respiratory failure) on a background of depletion of total body potassium. His initial magnesium was low normal at 0.66 mmol/L (reference range 0.6–1) and his initial phosphate was low at 0.35 mmol/L (reference range 0.7–1.5), both consistent with his chronic alcohol use.
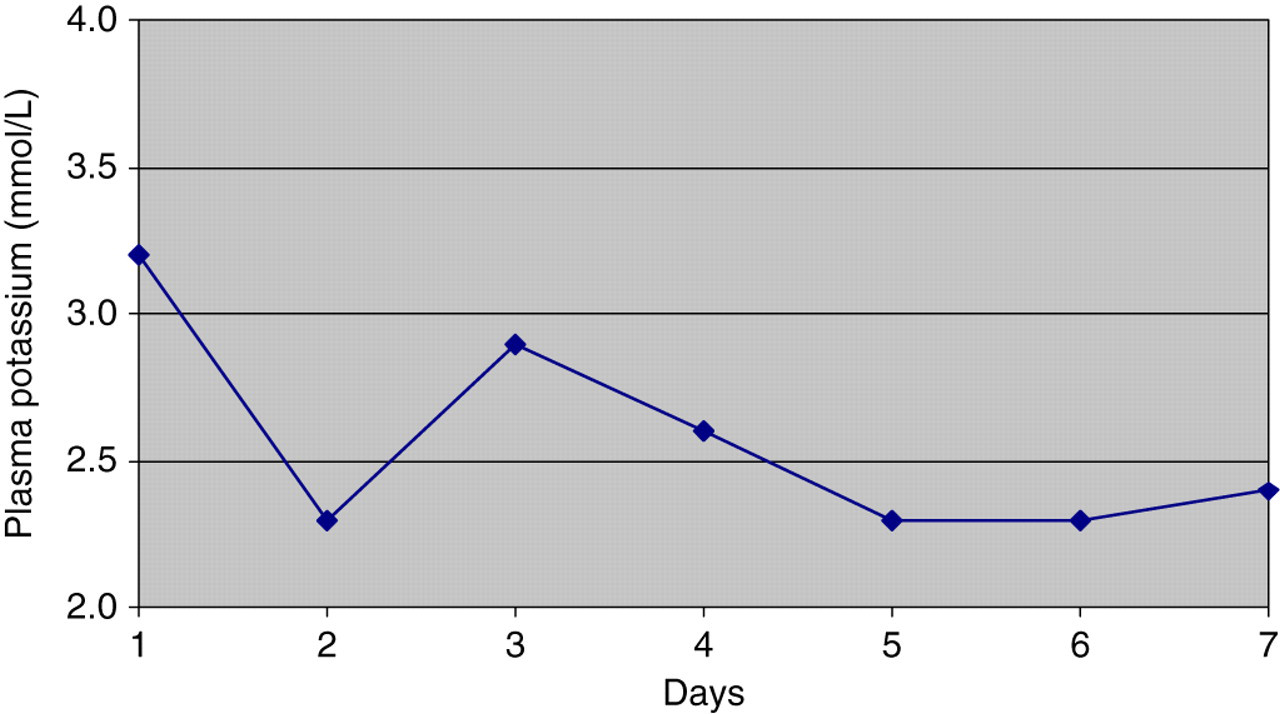
Potassium levels in first week following admission
Hypotonic polyuria (peaking at 6 L/24 h) developed alongside the reduction in plasma potassium. This in turn was followed by a slow rise in plasma sodium concentrations up to 161 mmol/L on day 7. The combination of the hypernatraemia and the hypotonic polyuria were consistent with the development of DI. Plasma glucose and ionized calcium were normal. The rise in plasma sodium did not exceed 12 mmol per 24 h at any time (Figure 2). A lack of response to intramuscular desmopressin (given to out-rule possible cranial DI) and subsequent correction of his polyuria and hypernatraemia immediately following normalization of plasma potassium confirmed the diagnosis of nephrogenic DI due to severe hypokalaemia. At this point recumbent aldosterone was measured and was <69 pmol/L (reference range 33–416) with a concomitant potassium of 3.7 mmol/L, and a recumbent renin of 4.3 ng/mL/h (reference range 0.5–2.6). His adrenocorticotrophic hormone concentration was 14.8 pg/mL (reference range 10–60), with a concomitant total serum cortisol of 205 nmol/L.
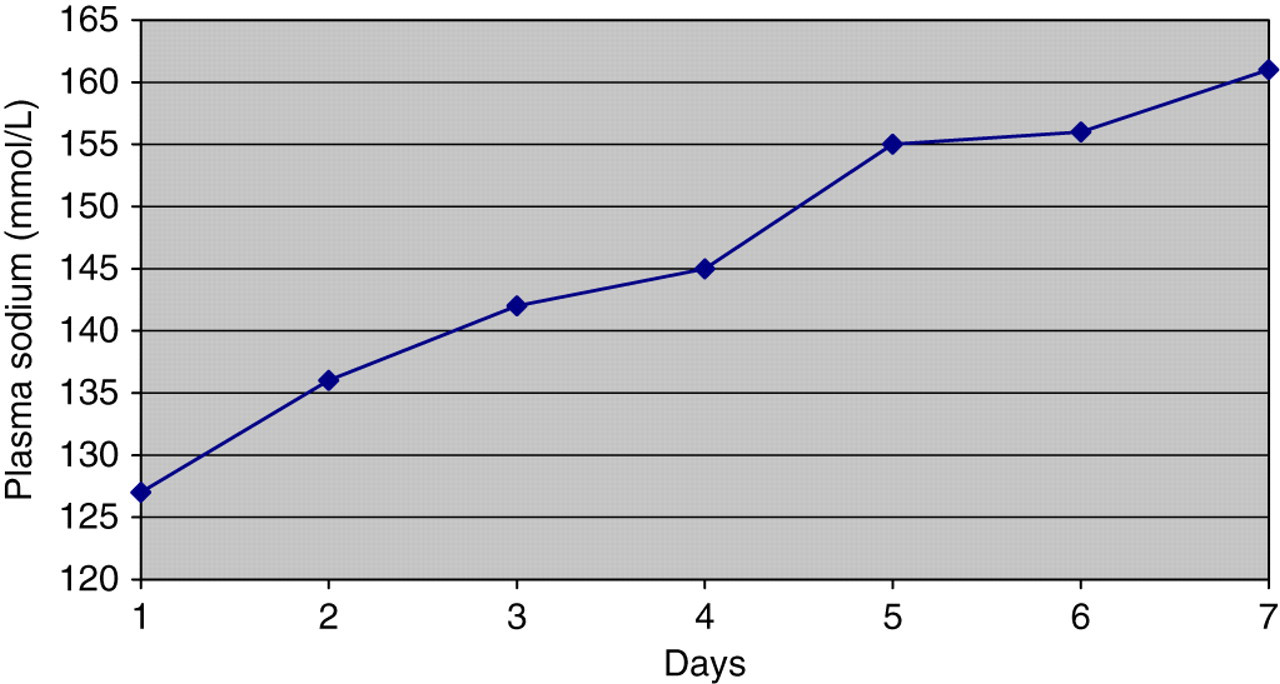
Sodium levels in first week following admission
During the first week, while the above electrolyte changes progressed, his respiratory status deteriorated with worsening hypoxaemia, hypercapnia and finally acidosis. He required mechanical ventilation in the intensive care unit (ICU), and under intensive management his electrolyte abnormalities and respiratory status slowly improved. He was weaned off sedation but remained obtunded with a Glasgow Coma Scale score of 3/15. On the advice of the endocrine team in attendance, MRI of the brain was performed, which confirmed the clinical suspicion of central pontine osmotic demyelination (Figure 3). The patient's condition did not improve and he finally died in ICU 10 days later.
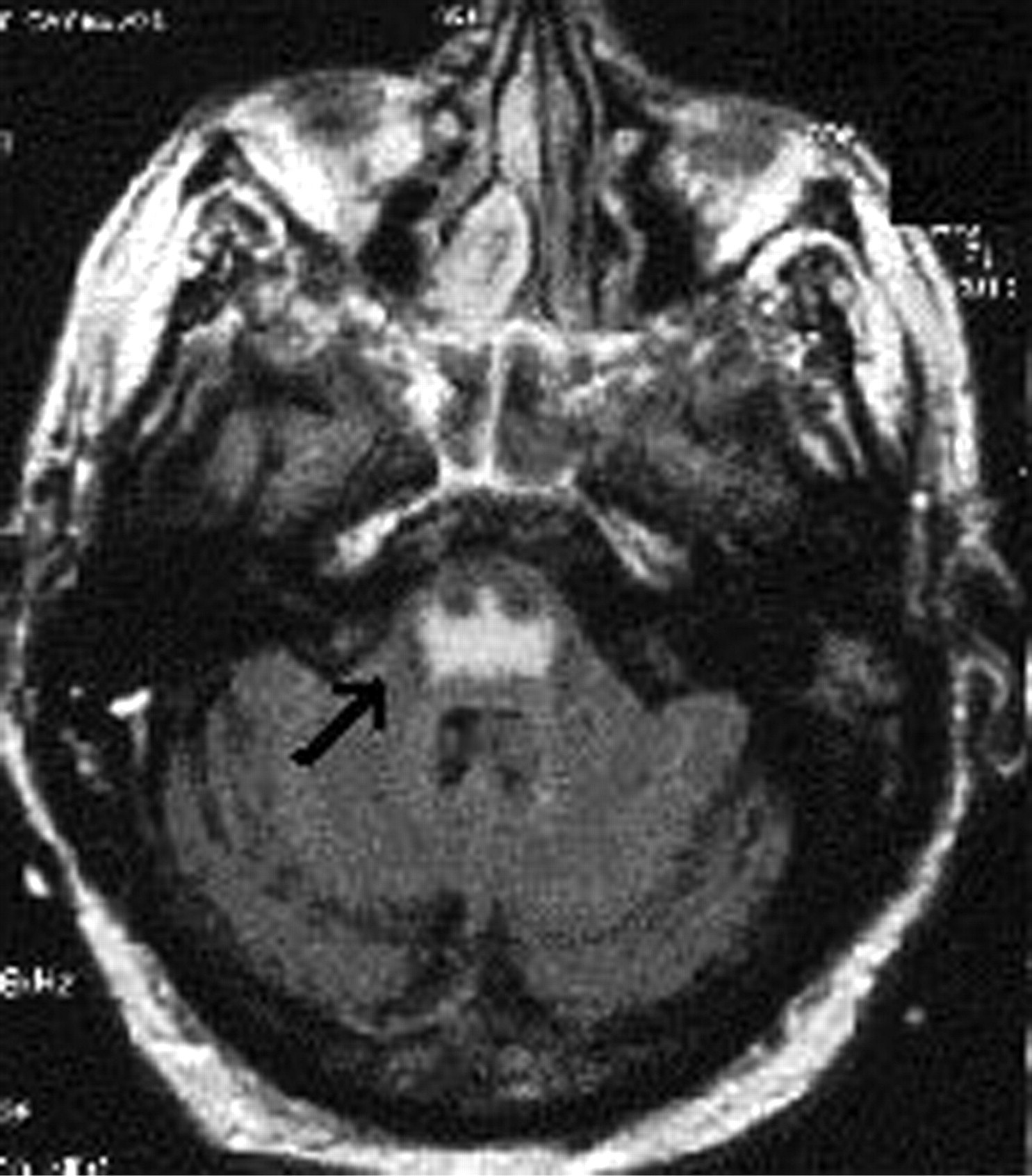
Bat-wings appearance characteristic of CPM (arrow)
Discussion
This is the first reported case of nephrogenic DI resulting in the development of CPM. The above clinical course is notable for several distinct points of interest. The first is the development of CPM despite a slow rise of plasma sodium that did not exceed recommended guidelines. The second is the domino effect of one electrolyte abnormality involving potassium depletion leading directly to another in which the initial hyponatraemia rapidly converted to hypernatraemia. The hypokalaemia is and of itself an additional independent risk factor for the development of CPM and hence in effect caused a double insult. Phosphate concentrations were initially low, which would be consistent with malnourishment from chronic alcohol use, but both magnesium and phosphate steadily increased from the point of admission and at no point did a precipitous drop in these electrolytes occur. This makes it unlikely that the refeeding syndrome was responsible for his hypokalaemia.
In addition to such classical causes as lithium treatment and hypercalcaemia, nephrogenic DI may be caused by severe and persistent hypokalaemia. 7 Oksche et al. 8 described reduced expression of the aquaporin-2 (AQP-2) channel on the renal collecting ducts secondary to low serum potassium concentrations. Diminished function of the AQP-2 receptor results in impaired sensitivity to antidiuretic hormone, and subsequent aquaresis. The nephrogenic DI results in dehydration and hypernatraemia in situations where water intake is inadequate to compensate for the polyuria such as in sick patients. The DI corrects with the normalization of serum potassium.
The present guidelines, which are aimed at minimizing the risk of CPM during the correction of chronic hyponatraemia (present for more than 48 h), recommend a rise in plasma sodium of not more than 12 mmol over 24 h 9–11 and not exceeding 25 mmol over 48 h. 12 In this patient's case significant osmolar injury developed despite a slow rise in plasma sodium that did not exceed 12 mmol/L/24 h from a baseline of relatively mild initial hyponatraemia, emphasizing the importance of additional variables in the development of CPM. Although the rise in plasma sodium in our patient was secondary to DI, nevertheless, this case queries the need for more caution in the more common situation of treating chronic hyponatraemia. 13 New guidelines may need to put greater focus on other risk factors for CPM such as potassium, hypoxia, nutritional status and history of alcohol excess as were present in our patient.
Chronic alcoholism remains the most common condition associated with osmotic demyelination, being present in 39% of reported cases since 1986. 2 It has been postulated that chronic alcohol abuse leads to depletion of glucose and glycogen in glial cells, diminishing their ability to operate their Na/K ATPase pump in the face of an osmotic challenge. 3 Correction of hyponatraemia is, in fact, the second most common co-associated condition being reported in 21.5% of cases. 2 CPM following the correction of hypernatraemia has also been documented, but far less frequently and often in the presence of hyperglycaemia. 4,14 Since the introduction of liver transplantation in the 1980s, this procedure has become the third most common co-associated condition (17%), with CPM typically occurring in the first 30 days post-transplant. 2 The aetiological link between liver transplant in particular and CPM has yet to be elucidated fully, although fluctuations in sodium, the onset of sepsis, the use of certain immunosuppressants and hypoxia have all been proposed as contributing factors in this population.
Less commonly reported than the above risk factors, but increasingly recognized as playing a role in the pathogenesis of CPM, is hypokalaemia. As far back as the seminal paper on CPM in 1959, Adams et al. 15 noted a possible role for potassium in the development of the disorder. More recently, Shintani et al. 16 have reported a case of CPM occurring in the absence of sodium flucuations, alcohol abuse or liver transplantation, but with significant hypokalaemia present. Heng et al. 5 in a prospective study of hyponatraemic patients entering ICU, noted that CPM occurred even when sodium was corrected slowly, but identified low baseline potassium as an independent risk factor for developing the condition. In our patients, hypokalaemia caused a double insult, resulting in hypernatraemia secondary to nephrogenic DI and also independently adding to the risk of CPM. Other conditions that have been reported as occurring with and potentially contributing to CPM in otherwise healthy patients include burn injuries, hypoxia, folate depletion, hypophosphataemia and lithium toxicity. 2
In recent years, in addition to the purely osmolar theory of myelinolysis, an autoimmune component to the syndrome has been theorized. 17 In essence, the fluctuations in osmolality that are associated with CPM have been shown to cause significant dehydration of brain endothelial cells. 18 This leads to disruption of the blood brain barrier and allows the entry of a variety of circulating immune factors to the central nervous system, where they can subsequently contribute to the demyelination process. Support for this theory comes from rodent studies in which dexamethasone exerted a protective effect against the development of myelinolysis, and human case reports in which patients with significant CPM responded to therapy with dexamethasone or intravenous immunoglobulin. 19–21
Ultimately, CPM arises from relatively acute and severe alterations in serum osmolality, which in turn arise most commonly from the correction of hyponatraemic states. However, a complex interaction occurs between fluctuating osmolality, the inherent ability of the glial and vascular cells to adapt, and possibly the patient's own immune system. Given the recurring presentations of CPM cases similar to our own in which sodium fluctuation was slow, current recommendations on correcting sodium may be inadequate if these other risk factors are present.
DECLARATIONS