
Abstract
We report a case of a 65-year-old lady who presented with acute confusion and profound hyponatraemia (plasma sodium of 97 mmol/L). Five years earlier she had developed sepsis and was found to have hyponatraemia, thought to be due to syndrome of inappropriate antidiuretic hormone secretion. The patient was lost to follow-up. The patient was covered with steroids and investigations confirmed primary adrenal failure with flat response of cortisol to adrenocorticotropic hormone (ACTH) stimulation and very high level of ACTH. Adrenal auto-antibodies were negative and a computed tomography of the adrenals showed bilateral adrenal calcifications, suggestive of previous haemorrhage or infarction. Bilateral adrenal calcification due to haemorrhage/infarction usually does not present with severe hyponatraemia; however, adrenal insufficiency should be excluded in all cases of severe hyponatraemia. In suspected cases, patients should be treated with steroids, even when symptoms or signs are absent, while results of investigations are awaited.
Introduction
Bilateral adrenal haemorrhage can occur in different clinical situations and may present in a multitude of ways. 1 The diagnosis should always be considered in all patients who have a risk factor to develop the condition. Since the advent of computed tomography (CT) scanning, bilateral adrenal haemorrhage is increasingly recognized as a cause for adrenal insufficiency in adults. The unique vascular supply of the adrenal gland makes it susceptible for haemorrhagic necrosis. 2 Predisposing factors include sepsis, prothrombotic states such as the antiphospholipid syndrome and hypocoagulant states such as heparin diathesis and stressful events such as surgery. 2
Bilateral adrenal haemorrhage with subsequent infarction rarely presents with hyponatraemia. 2 Here we present an unusual case of profound hyponatraemia, which was proven to be due to primary adrenocortical failure secondary to previous haemorrhage. The salient features of the case and implications for clinical practice will be highlighted.
Case report
A previously fit and healthy 65-year-old lady was referred by her general practitioner in 2003 with a day's history of right-sided headache and drowsiness as well as several bouts of vomiting and confusion. At presentation she was normotensive, blood pressure (BP) was 130/50 mmHg, with a radial pulse of 100 bpm, pyrexial with a temperature of 39.4°C, and had neck stiffness, photophobia as well as exudates on both tonsillar fauces. The rest of the clinical examination was unremarkable. Initial investigations showed hyponatraemia with a serum sodium of 121 mmol/L (135–145 mmol/L), a potassium of 3.6 mmol/L (3.5–5.0 mmol/L), normal urea and creatinine. Full blood count showed a haemoglobin of 13.5 g/dL (11.5–16.4 g/dL), a white cell count (WCC) of 13.3 × 109/L (4–11.5 × 109/L), with neutrophil leukocytosis, but no eosinophilia, and a platelet count of 234 × 109/L (150–450 × 109/L). She also had random blood glucose of 5.6 mmol/L, C-reactive protein (CRP) 202 mg/L (2–10 mg/dL), negative blood cultures and throat swabs. Cerebrospinal fluid (CSF) obtained from lumbar puncture was clear and colourless. There was no xanthochromia, and the CSF cell count of 1 × 106/L, glucose 2.9 mmol/L (2.5–3.9 mmol/L) (corresponding blood glucose of 4.2 mmol/L), protein 0.20 g/L (0–0.45 g/L), and no organisms seen on Gram stain and no growth from cultures even after enrichment. The patient's haemoglobin dropped to 11.9 g/dL the day after admission, but that was attributed to haemodilution. Hyponatraemia was initially attributed to possible syndrome of inappropriate antidiuretic hormone secretion secondary to meningitis. Meningitis, however, was excluded and she was discharged home a few days later with a diagnosis of sepsis due tonsillitis. The patient was lost to follow-up.
In May 2008, she was admitted electively by the orthopaedic surgeons for a left total knee replacement. In the preoperative assessment her sodium was 129 mmol/L, po-tassium 4.5 mmol/L, urea 5.5 mmol/L, creatinine 62 μmol/L, glucose 4.4 mmol/L, haemoglobin 11 g/dL and mean cell volume (MCV) 88 fl, platelets 308 × 109/L, WCC 4.2 × 109/L. The mild hyponatraemia was overlooked, and the operation was uneventful; however, postoperatively her sodium dropped to 125 mmol/L, and this was attributed to intravenous fluids given and she was discharged home five days postoperatively. Two weeks later, she was readmitted to hospital with a five-day history of nausea, vomiting and confusion. The hyponatraemia had worsened, sodium being only 97 mmol/L, potassium 3.9 mmol/L, haemoglobin 9.3 g/dL, CRP < 5 mg/L and blood glucose 5.1 mmol/L. She was normotensive with a BP of 140/80 mmHg, apyrexial with a temperature of 36.2°C, Glasgow Coma Scale of 13/15 and an otherwise normal examination. No hyperpigmentation was noted. Plasma osmolality was low at 205 mosm/kg (280–300 mosm/kg), urine osmolality 424 mosm/kg, urine sodium 61 mmol/L, and a random cortisol and short synacthen test were done, and she was started on intravenous hydrocortisone (100 mg qds) pending the result of the short synacthen test. The patient had a dramatic response with significant improvement in her clinical status and gradual improvement of her sodium level, rising to 105 mmol/L after one day, and to 117 mmol/L after three days. Random cortisol level came back very low for the degree of her stress, at 174 nmol/L, and a short synacthen test showed a flat response with a 30 min cortisol value of 188 nmol/L, 60 min value of only 199 nmol/L, and adrenocorticotropic hormone (ACTH) level was very high at 162 mU/L (normal <20 mU/L). Other hormone levels were all normal including prolactin 102 mU/L (1–650 mU/L), thyroid-stimulating was hormone 0.64 mU/L (0.3–6 mU/L), and Free T4 20.7 pmol/L (9–24 pmol/L), luteinizing hormone 21.8 U/L, follicle-stimulating hormone 36.3 U/L, parathyroid hormone 5 pmol/L. However, her adrenal androgens were very low with dehyroepiandrostenedione sulphate <0.8 μmol/L, androstenedione <1.4 nmol/L, and testosterone <0.5 nmol/L (1.0–3.2 nmol/L). Sex hormone binding globulin was 47 nmol/L. Coagulation screen was normal, and full thrombophilia screen including genetic studies, anticardiolipin and antilupus antibodies were negative. Adrenal autoantibodies were negative. A CT brain scan showed no significant abnormality and a CT scan of adrenals showed bilateral adrenal calcification and atrophy, indicative of previous haemorrhage (Figures 1 and 2). No abnormality was detected on chest X-ray.
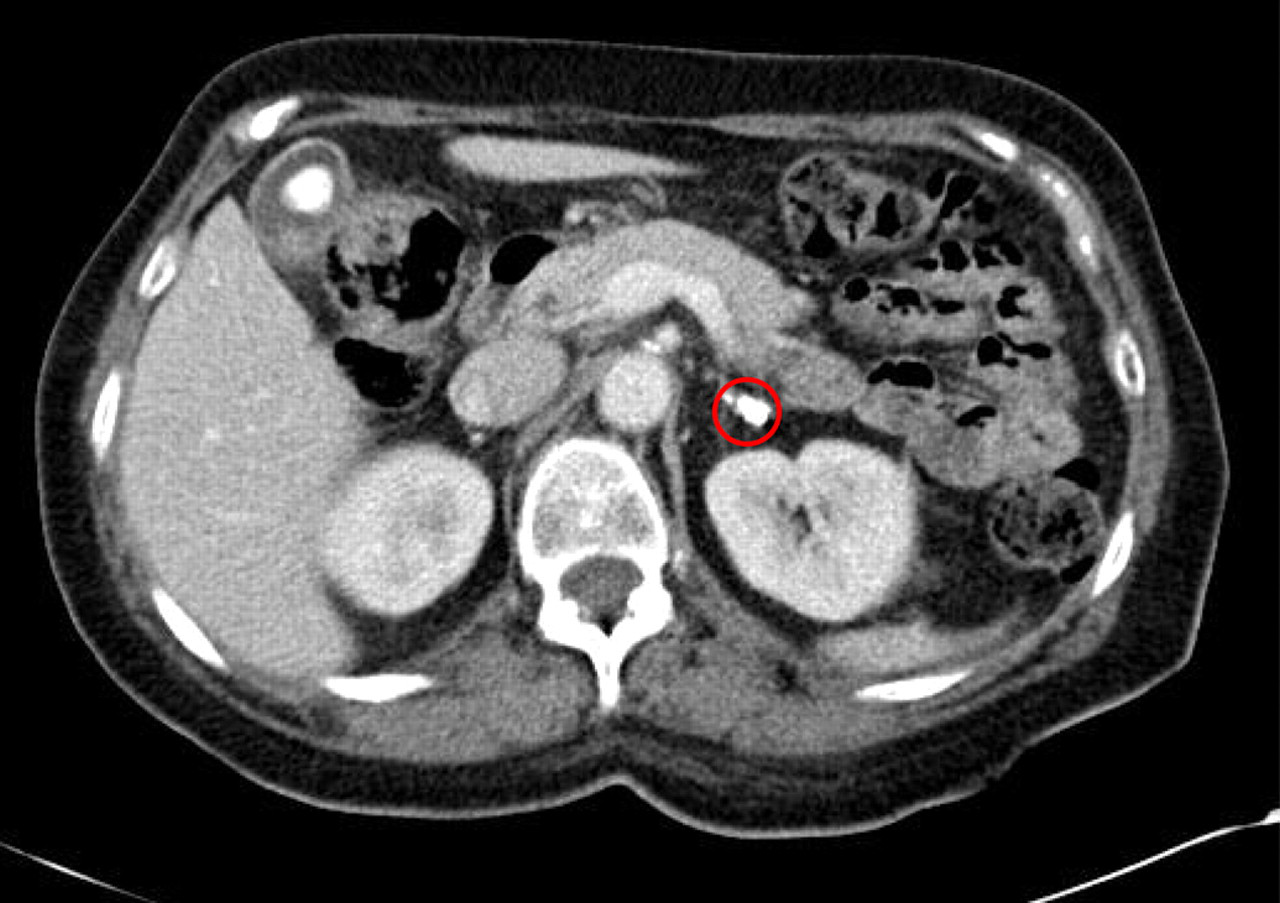
Bilateral adrenal calcifications of the adrenals, coronal view
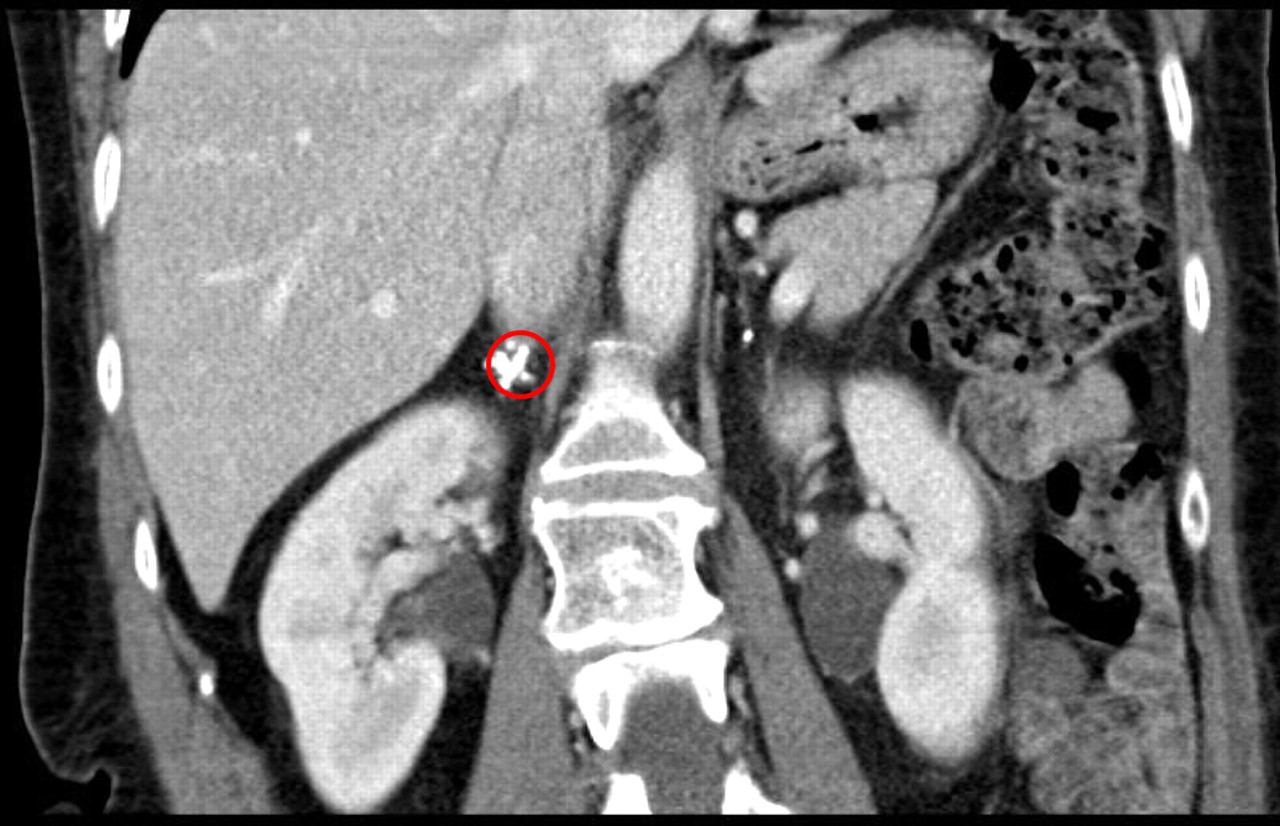
Bilateral adrenal calcifications of the adrenals, sagittal view
The patient continued to improve clinically and biochemically, with sodium levels rising gradually to normal. She was discharged home after 17 days with a plasma sodium of 136 mmol/L, and potassium 4.3 mmol/L on oral hydrocortisone 20 mg (am) and 10 mg (pm). There was no clinical evidence of mineralocorticoid deficiency so fludrocortisone replacement was not required. On regular follow-up she remains well with no further episodes of confusion and hydrocortisone was decreased to 20 mg per day.
Discussion
Adrenal haemorrhage, with subsequent infarction of adrenal tissues, usually presents abruptly with severe hypotension and collapse. Sometimes, as in our case it may present insidiously, making it difficult to make a correct diagnosis. Adrenal insufficiency caused by bilateral adrenal haemorrhage rarely presents with hyponatraemia, and profound hyponatraemia is indeed a very unusual presentation. In a study on non-traumatic adrenal haemorrhage by Vella et al. 2 at the Mayo Clinic, only 9% had hyponatraemia (sodium <130 mmol/L) and 16% had hyperkalaemia (potassium >5 mmol/L). In this series a pronounced decrease in haemoglobin (>1.5 g/dL) was common in those who experienced spontaneous adrenal haemorrhage, which was often associated with massive retroperitoneal haemorrhage. Our patient had a drop in her haemoglobin at the time of her initial admission with septic illness. Thrombocytopenia and prolonged activated partial thromboplastin time were common in patients with antiphospholipid syndrome, but also in those patients whose adrenal haemorrhage is caused by sepsis or stress. 2 Our patient presented with acute on chronic decline in her sodium, which reached a nadir after five years and her presentation culminated as profound hyponatraemia with intense organic brain syndrome. Furthermore, the clinical and biochemical abnormalities in our patient were not typical of primary adrenocortical failure with the absence of hyperkalaemia, hypotension and hyperpigmentation despite the long history of her hyponatraemia. We believe that the haemorrhage of the adrenals occurred in the process of her initial septic illness in 2003, but her adrenal failure was not complete and this progressed over the years. She was noted by her daughter to be ‘craving salt’ for years and this (as well as the residual mineralocorticoid function) is what kept her sodium and blood pressure within reasonable levels. It is likely that the stress of the knee operation led to her acute adrenal crises.
In about 50% of cases, bilateral adrenal haemorrhage is associated with an acute illness (e.g. sepsis, congestive heart failure, myocardial infarction, complications of pregnancy) or an event (e.g. surgery or an invasive procedure). 3 Other frequent associations include haemorrhagic diatheses (e.g. anticoagulant use, thrombocytopenia), thromboembolic disease, including antiphospholipid syndrome, blunt trauma and ACTH therapy (used in the past to treat patients with inflammatory bowel disease). 4–10 The strong association between non-traumatic adrenal haemorrhage with haemorrhagic diathesis or coagulopathy suggests that haemorrhage may be a primary event and is not initiated by thrombosis. In a patient with thrombotic disease, adrenal vein thrombosis may occur first and subsequent anticoagulant therapy may precipitate haemorrhage. 11 Presenting symptoms and signs of bilateral adrenal haemorrhage are usually non-specific, and include abdominal pain (56%), back or flank pain (21%), chest pain (13%), nausea and vomiting (46%), fever (59%), hypotension/shock (74%) and tachycardia (28%) 1,2 Although the precise mechanism(s) leading to adrenal haemorrhage are unclear in non-traumatic cases, available evidence has implicated ACTH, 7–9 adrenal vein spasm and thrombosis, and the normally limited venous drainage of the adrenal in the pathogenesis of this condition. The surge in ACTH with the stress of septic illness may be the trigger in susceptible patients. The central adrenal vein has a unique arrangement in that its musculature is eccentric and is composed of thick, longitudinal muscle bundles. Turbulence and local stasis may occur in this vein when the bundles contract; the result is venous thrombosis and haemorrhagic infarction. 2 For instance, in a patient with a hypercoagulable state secondary to antiphospholipid syndrome, adrenal vein thrombosis is believed to be the cause of haemorrhagic infarction. 11 Our patient has no evidence of coagulopathy or thrombophilia on testing, and the most likely culprit is the surge of ACTH with her initial septic illness.
CT scanning is the investigation of choice for diagnosing adrenal haemorrhage in adults. Adrenal calcification on CT scanning is most commonly caused by haemorrhage, 12 although there are other causes for adrenal calcification and this may on its own be an incidental finding. 13 Tuberculous adrenalitis was also a major cause of adrenal calcification in the past, 14 but nowadays with the resurgence of cases of tuberculosis (TB), especially in the immunosuppresed, the diagnosis of TB adrenalitis should also be considered when adrenal calcification is present. The involvement of the adrenals with TB is not usually an isolated event in cases of TB, and the patient would normally have evidence of TB elsewhere, and would be very unwell with systemic disease. Our patient had no past history of TB, had normal chest X-ray, nor was there any evidence to suggest systemic TB in her case, to justify obtaining a biopsy from the calcified adrenals. Furthermore despite the fact that it would be extremely difficult to prove that this is not tuberculous adrenalitis, the clinical evidence is arguing against TB as a cause of her bilateral adrenal calcifications.
Despite the plethora of information about hyponatraemia in the hospital settings and the advent for proper investigations and management of these cases, however, hyponatraemia continues to be challenging, and unusual aetiologies and atypical presentation continue to emerge, necessitating high level of awareness among clinicians. 15–17 All of these studies have shown that only few patients with hyponatraemia get their cortisol checked, and fewer patients get their adrenal function fully assessed. One must always exclude adrenal insufficiency as a cause of profound hyponatraemia especially in patients with risk factors for adrenal haemorrhage, such as postoperative patients, those on anticoagulants or those suffering from septicaemia. Furthermore, patients with severe hyponatraemia should be promptly covered with potentially life-saving steroids after taking blood samples for cortisol.
DECLARATIONS