
Abstract
Aims
Current screening methods, such as single strand conformational polymorphism (SSCP) and denaturing high performance liquid chromatography (dHPLC) that are used for detecting mutations in familial hypercholesterolaemia (FH) subjects are time consuming, costly and only 80–90% sensitive. Here we have tested high-resolution melt (HRM) analysis for mutation detection using the Rotor-Gene6000 realtime rotary analyser.
Methods and subjects
Polymerase chain reaction and melt conditions (HRM) for 23 fragments of the LDL-receptor gene, a region of exon 26 in the APOB gene (including p.R3527Q) and exon 7 of the PCSK9 gene (including p.D374Y) were optimized. Two double stranded DNA saturating dyes, LC-Green and Syto9, were compared for sensitivity. Eighty-two samples with known mutations were used as positive controls. Twenty-eight Greek FH heterozygous patients and two homozygous patients from the UK and Croatia were screened.
Results
HRM was able to identify all the positive control mutations tested, with similar results with either dye. Eight different variations were found in 17 of the 28 Greek FH patients for an overall detection rate of 61%: c.41delT (1), p.W165X (1), p.C173R (3), p.S286R (2), p.V429M (4), p.G549D (4), p.V613I (1), and a previously unreported mutation p.F694V (1) which is predicted to be FH-causing by functional algorithms. Mutations were found in both the homozygous patients; p.Q92X (Croatia) and p.Y489C (UK); both patients were homozygous for their respective mutations.
Conclusions
HRM is a sensitive, robust technique that could significantly reduce the time and cost of screening for mutations in a clinical setting.
Introduction
Familial hypercholesterolaemia (FH) is an autosomal dominant disorder associated with elevated levels of plasma low-density lipoprotein (LDL) particles and increased risk of coronary heart disease (CHD), 1,2 with an estimated prevalence in the UK of one in 500 to one in 600. 3 Mutations in the LDL-receptor gene (LDLR) account for the majority of identified mutations 1,2 and to date, more than 1000 LDLR mutations have been reported in FH patients worldwide, with more than 200 different mutations found in UK patients. 4 The LDL particle contains one molecule of the apolipoprotein apoB, which is the ligand for the LDL receptor, and in patients in the UK with a clinical diagnosis of FH about 5% carry one particular mutation (p.R3527Q) in the APOB gene. 5 Defects in the protein convertase subtilisin/kexin type 9 (PCSK9) gene can also cause monogenic hypercholesterolaemia. 6 The protein is involved in the degradation of the LDL-receptor protein in the lysosome of the cell and prevents it recycling (see references in ref 7 ). Gain of function mutations in the PCSK9 gene therefore cause increased degradation of LDL-receptors, reduced numbers of receptors on the surface of the cell and monogenic hypercholesterolaemia. One mutation in this gene, p.D374Y, occurs in about 2% of UK FH patients 8 and is associated with particularly early onset of CHD. 9
The efficacy of molecular genetic testing for FH is dependent on both the sensitivity and specificity of methods of mutation detection, with current rates of detection ranging from 50% to 90%. Screening methods, such as SSCP 10 and dHPLC, 11 used for detecting mutations in FH subjects are time consuming, costly and only 80–90% sensitive. 12 Testing for known mutations by methods such as a Chip, 13 ARMs 14 or MassARRAY spectrometry (iPLEX) 15 have utility, but clearly cannot test for previously unreported variants, while sequencing, 16 which remains the gold standard, is still costly and relatively time consuming. Currently, test results may take several months to be reported and costs for this are in the range of £500–£1000 (∼50% cost is for labour). 17 In the current study, we evaluate the use of high-resolution melt (HRM) analysis for mutation detection as a rapid screening tool for FH patients. HRM curve analysis is a simple and cost-effective post-polymerase chain reaction (PCR) technique, which is used for high throughput mutation scanning. 18,19 It characterizes nucleic acid samples based on their disassociation (melting) behaviour. Samples can be discriminated according to their length, sequence, GC content or strand complementarity. Single base changes can be readily identified. This is a closed tube method that requires no post-PCR handling such as gel separation, and therefore may improve analysis time. The technique requires the use of standard PCR reagents and a saturating double-stranded (dsDNA) binding dye such as LC-Green or Syto9. The process of HRM relies on the presence of these saturating dsDNA binding dyes that have the ability to register the smallest degree of melting from double- to single-stranded DNA by a change in fluorescence signal intensity. The technique has already been employed to scan for somatic mutations in the TP53 gene 18 and the APOB gene, 19 and after submission of this paper a report of its use for the LDLR has also appeared. 20
Materials and methods
Patient selection criteria
Twenty-eight Greek FH heterozygous patients and two homozygous patients from the UK and Croatia, with DNA from the parents of the Croatian proband, were screened. 21 The proband from Croatia was identified at the age of three years with clinical signs of homozygous FH (elevated LDL-cholesterol levels and xanthomas). Heterozygous hypercholesterolaemia was found in his parents and some other family members. The boy has been treated with simvastatin and atorvastatin, but without reaching the treatment goals. The UK patient is of Pakistani origin, who on referral at the age of 14 y for juvenile rheumatoid arthritis was noted to have some lipomatous skin lesions. She was only referred to the Lipid Clinic in Buckinghamshire in 2007 with recorded total cholesterol in the referral letter of 20 mmol/L. After one month treatment with simvastatin, total cholesterol level was 15.7 mmol/L. Her father died at the age of 64 y from ischaemic heart disease (IHD), but was reported to have suffered from angina from the age of 57 y, and was known to have suffered from type 2 diabetes and had elevated cholesterol levels (9.1 mmol/L). Her mother (born 1937) has suffered from hypertension since 2002 and was diagnosed with diabetes in 2005. Her parents are first cousins. The proband has two sons aged 4.5 y and five months and a daughter aged two years. Her current treatment is with 40 mg Rosuvastatin, 10 mg Ezetemibe and 2 g plant sterols per day, and lipid levels at her latest clinic visit are shown in Table 1. Standard assays for plasma lipids were performed as described 21,22 LDL concentrations were calculated from fasting lipid samples using the Friedewald formula. Patients were designated using the UK Simon Broome criteria, 1,23 as Definite FH or Possible FH on the basis of the presence or absence of tendon xanthomas. All patients gave their informed consent and the ethical committee of Harokopio University approved the study.
Baseline characteristics of the homozygous FH probands and available relatives
LDL-C, low-density lipoprotein-cholesterol; HDL-C, high-density lipoprotein-cholesterol; CHD, coronary heart disease
*F = Female, M = Male
†on treatment 40 mg Rosuvastatin, 10 mg Ezetimibe, 2 g plant sterols
Molecular genetic analysis
Polymerase chain reaction and high-resolution melt
Genomic DNA was isolated from whole blood samples using standard methods. Oligonucleotide primers for PCR–HRM were designed to cover, where appropriate, intron–exon junctions and up to 60 bp of the introns (Supplementary Table 1). The promoter and coding regions of the LDLR together with the p.R3527Q mutation (part of exon 26) of APOB and p.D374Y (exon 7) of the PCSK9 gene were included for screening. PCR and subsequent HRM were carried out in the Rotor-Gene6000 (Qiagen Ltd, Crawley, West Sussex, UK). Exon 10 of the LDLR was screened by two overlapping PCR fragments and exon 4 by four overlapping fragments. PCR for the most 3′ fragment of exon 4, designated as e4(4), was performed on the G-Storm GS4 thermal cycler (GRI) using 20 µL reactions (2 mmmol/L MgCl2; PCR mix as below). Eight microlitres of this PCR were transferred to Rotor-gene tubes and 2 µL of Syto9 (Invitrogen Ltd, Paisley, UK) added to give a final concentration of 1.5 µmmol/L. The PCR/Syto9 mix was first denatured at 96/2 min and then cooled to 25/2 min on the Rotor-Gene6000 before HRM was carried out. Comparison of sensitivity and cost of LC-Green and Syto9 dyes was also carried out while optimizing the PCR fragments. The 25 PCR–HRM fragments were optimized using wild-type DNA samples at four MgCl2 concentrations (2 mmmol/L, 3 mmmol/L, 4 mmmol/L, 5 mmmol/L for Syto9 and 3 mmmol/L, 4 mmmol/L, 5 mmmol/L, 6 mmmol/L for LC-Green) and three annealing temperatures of 57, 62 and 67°C. Conditions for PCR (10 µL reactions) were; KCl 50 mmmol/L, Tris 10 mmmol/L, gelatin 0.001%, dNTPs (each) 2 mmmol/L, MgCl2 (as appropriate), 0.4 units Taq DNA polymerase (Invitrogen) plus Syto9 to 1.5 µmmol/L or LC-Green to a 1× solution. Optimization was considered to be the conditions under which wild-type samples showed the least spread in melting; the tightest HRM curve, against having sufficient PCR product to give a good melt. After optimization with wild-type samples the PCR/HRM was repeated under optimum conditions with wild-type and positive control mutation samples to ensure detection of all known mutations.
Positive controls used to optimize HRM for the LDLR
HRM, high-resolution melt
*Variants in italics are common polymorphisms also detected by HRM
The enzymes used to detect them are shown, together with the rare allele frequency
†Forced site PCR
‡T726I is a rare variant (carrier frequency ∼1%) which is not FH causing
Mutations were designated according to
Homozygous patients
In addition to the initial round of screening, DNA from the homozygous patients was re-screened by HRM after adding wild-type PCR for each of the fragments. PCR (10 µL) and HRM were performed as previously described for wild-type and homozygous patients separately. Wild-type PCR product, 5 µL, was then added to the homozygous patients PCR, 5 µL, and the combined products heated to 96°C for two minutes to denature them and then cooled to 25°C for two minutes to re-anneal before repeating the HRM. Both the homozygous sample and the wild-type sample were matched for common LDLR single nucleotide polymorphisms (SNPs) when applicable (e.g. in exons 10, 12, 13, etc.), amplified and HRM performed before mixing as well as after mixing.
Restriction fragment length polymorphism analysis
Common SNPs in the LDLR were analysed as previously described 10 using enzymes as shown in Supplementary Table 2.
Primers used to amplify fragments for HRM
Sequencing
PCR products that revealed potentially FH-causing HRM melt profiles were subsequently sequenced using the MegaBACE-1000, 96 capillaries DNA sequencing system (GE Healthcare Life Sciences, Little Chalfont, Buckinghamshire, UK) or ABI-377 sequencing system (Applied Biosystems Inc, CA 94404, USA). Sequencing was carried out using fresh PCR products or directly from the HRM–PCR. Detected variants were designated as pathogenic or non-pathogenic using criteria as previously described.
24
Mutation nucleotide numbers were designated using the LDLR sequence reported (
Results
Optimizing conditions
HRM runs for all 25 fragments with 81 known FH heterozygous mutation-positive controls (Supplementary Table 1) and non-mutation ‘wild-type’ samples showed that HRM was able to detect a different melt profile from wild type for all of the tested mutations, with repeatable results on subsequent runs. All the fragments were optimized using both Syto9 and LC-Green dyes; LC-Green always required a slightly higher magnesium concentration (not shown). Both dyes were able to distinguish the known positive mutation controls, although LC-Green gave slightly tighter melt curves in many cases (i.e. less within-run variability, not shown).
Figure 1 shows a typical HRM melt profile for exon 3 with 50 wild-type samples (in red) and nine with a known exon 3 mutation. For the normal samples the differences between traces are small, showing good reproducibility. All the melts from subjects with known mutations (in blue) gave a curve that was clearly distinct from the normal, showing that the method and conditions used were working well. It was not possible to identify unambiguously one mutation melt from another, that is, the mutation melts are not unique, though some are distinct (e.g. p.V66fs). The APOB and PCSK9 fragments also gave a clear difference in HRM from the wild type (see the inset in Figure 3).
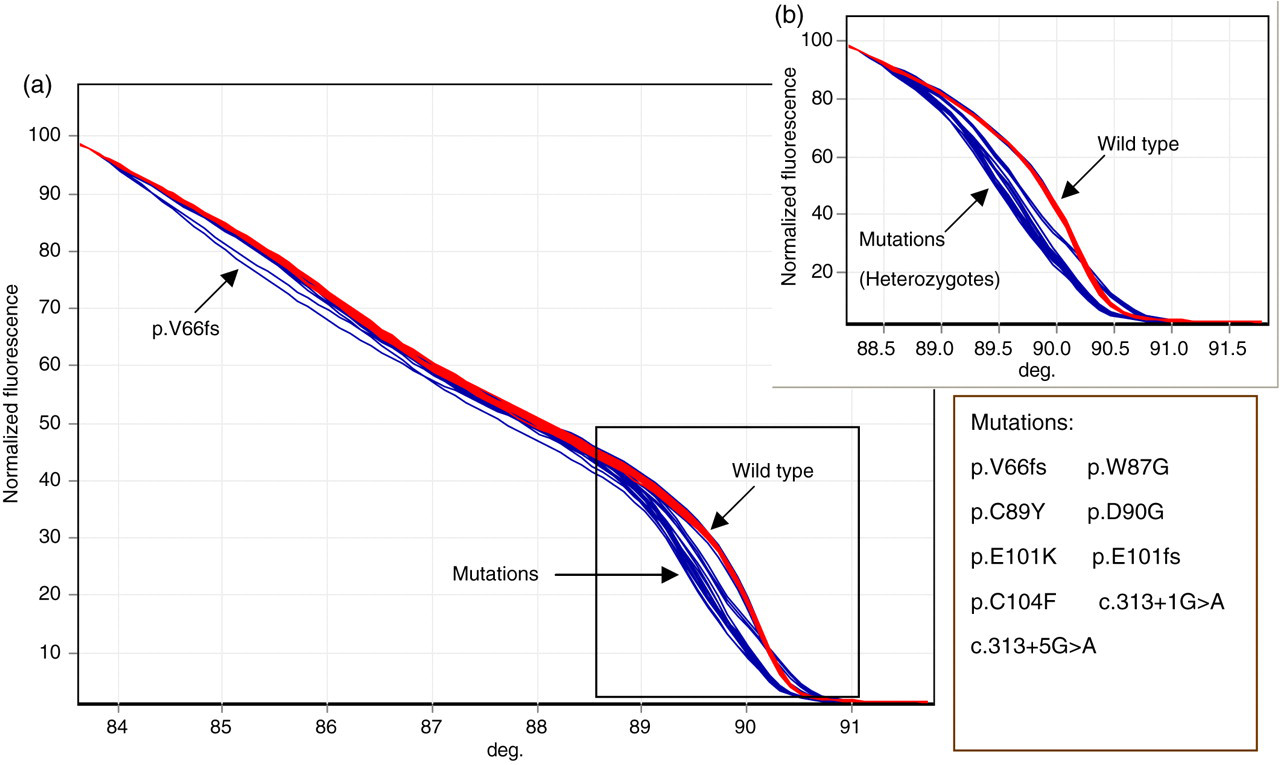
HRM of exon 3 of 50 wild-type samples and 10 mutation-positive samples (in duplicate). Wild-type curves shown in red; mutations in blue. (a) Showing the melt from 84 to 91°C. FsV45 is only detectable between 85 and 87°C. (b) Enlargement of the melt between 89.5°C and 91°C. FsV45 cannot be distinguished in this temperature range, as it is similar to the wild-type melt profile
We were unable to optimize the PCR for the exon 4 3′ (ex4(4)) fragment with Syto9 or LC-green on the Rotor-gene, and PCR for this fragment was thus performed on a separate thermal cycler and samples prepared for HRM as described above. As shown in Supplementary Figure 1, the ‘spread’ of curves for the wild-type samples was larger than for the standard run, but the two tested mutations gave a distinct melt curve, and this method is an acceptable alternative to PCR on the Rotor-gene.
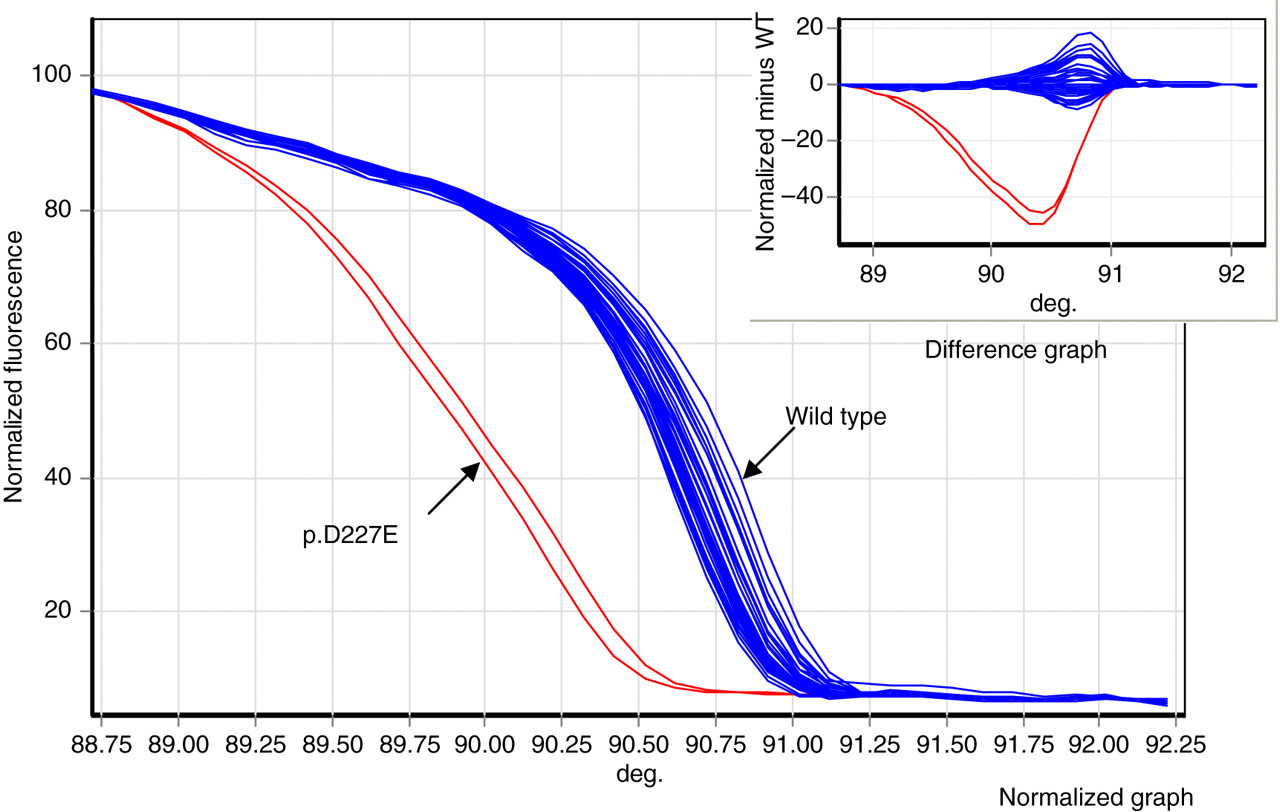
HRM Run IV for LDLR fragment ex4(4) in 59 wild-type samples and a mutation positive control. The 3′ most fragment of exon 4, ex4(4), PCR was performed in the G-Storm thermal cycler and then transferred to Rotor-gene tubes and dsDNA saturating dye, Syto9, added. The whole was then denatured and cooled before HRM was performed. The figure shows the normalized and difference graphs for the wild-type and mutation control (p.D227E, in duplicate)
Polymorphisms
The amplified fragments of the LDLR include several common SNPs that also generate variant HRM patterns. Figure 2 shows the pattern obtained with exon 13 which has a common polymorphism. To overcome this, in a single-fragment 72 sample run, the samples were genotyped by restriction fragment length polymorphism (RFLP) methods and the different genotypes analysed separately using the HRM software. Any variant from the genotype curve was then sequenced. Since the dsDNA binding dyes do not interfere with restriction enzyme digest, samples were genotyped for the polymorphism by RFLP using the PCR from the HRM run, or a fresh PCR. A typical example of this is shown in Figure 2 for the exon 13 C > T polymorphism (rare allele frequency 0.49). In Figure 2a, showing the normalized HRM graph, the three polymorphism genotypes are easily distinguished and the variant melt profile of two of the mutations is evident, but the melt for p.W599C is not clearly distinguishable. In Figure 2b the difference plot (subtracting the value for a single CT-heterozygote) is shown. Again, all three genotypes are distinct and the patterns for all mutations including p.W599C are now easily distinguished.
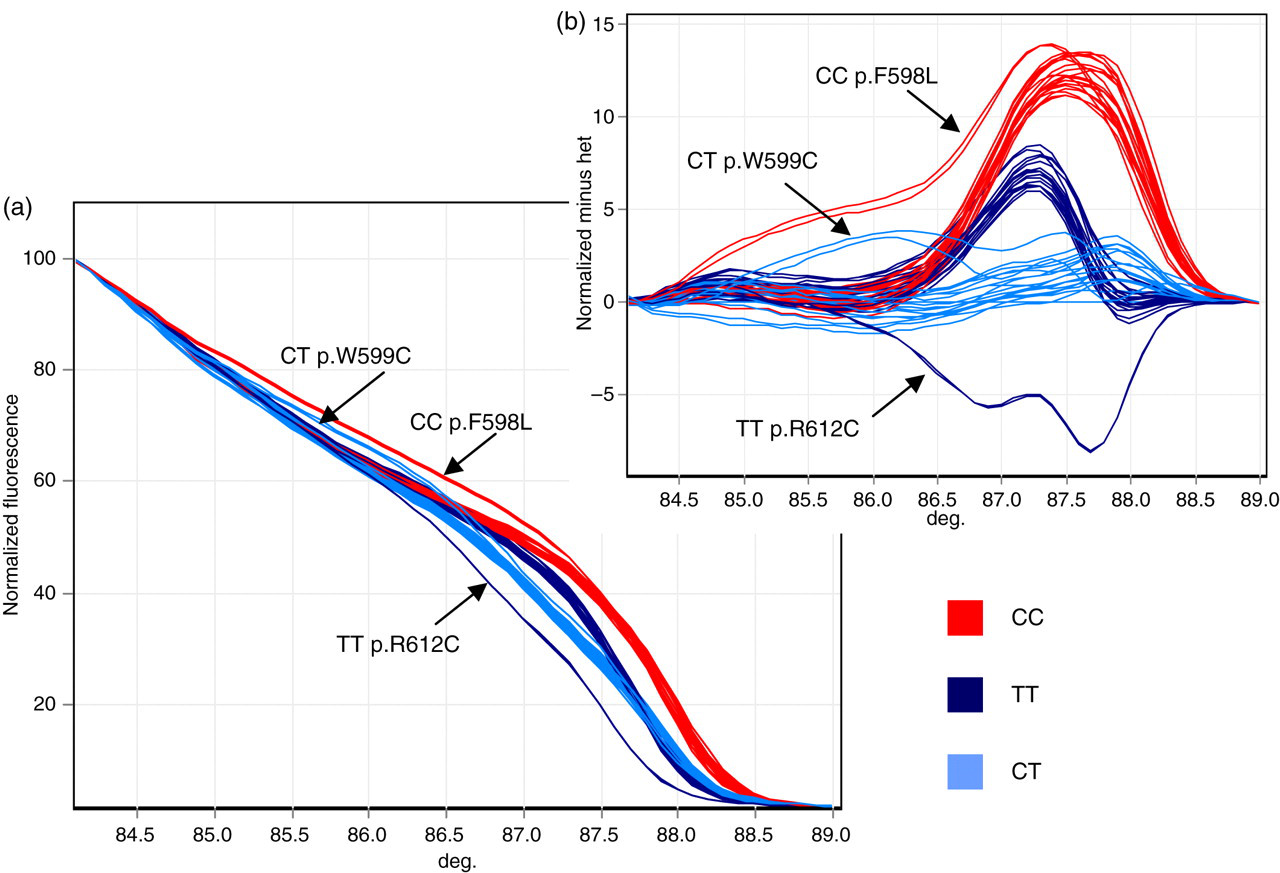
HRM of exon 13 showing the melt profile of the three genotypes of the common polymorphism and three mutations. For the Exon 13 C > T polymorphism (rare allele frequency 0.49), CC homozygotes are shown in red; CT heterozygotes in blue; TT homozygotes in dark blue. (a) Normalized HRM graph showing exon 13 melt with mutation-positive controls. The three genotypes are easily distinguished. The genotypes of the mutations are shown. The p.W599C is not clearly distinguishable. (b) Difference plot of the normalized HRM with values from the averaged CT-heterozygote subtracted. All three genotypes are distinct and the patterns for all mutations including p.W599C are easily distinguished
Testing new FH samples
As well as performing single exon runs we have determined the feasibility of running PCR/HRM on the rotor gene for screening a few patient samples for several fragments in one run. A method for covering all 25 fragments in four runs for eight patient samples was devised. Different ways of combining samples and machine runs were carried out to develop a final protocol as time efficient as possible. The resulting combinations are shown in Table 2. The results from Run II are shown in Figure 3, where exons 2, 12, 14, 15, 18 of the LDLR, exon 7 of PCSK9 covering p.D374 and part of exon 26 of APOB covering p.R3527 are run simultaneously. Exon 14 shows a mutation (subsequently determined to be p.F694V), while exons 15, 2 and 12 show polymorphism profiles. No other exons show a variant melt, and neither does the APOB or PCSK9 fragment. Using this protocol all the 25 fragments can be screened for eight patients in four PCR/HRM runs. This can be achieved in one working day. In the combined fragment protocol samples showing a variant pattern, even within an exon with a known polymorphism, were sequenced.
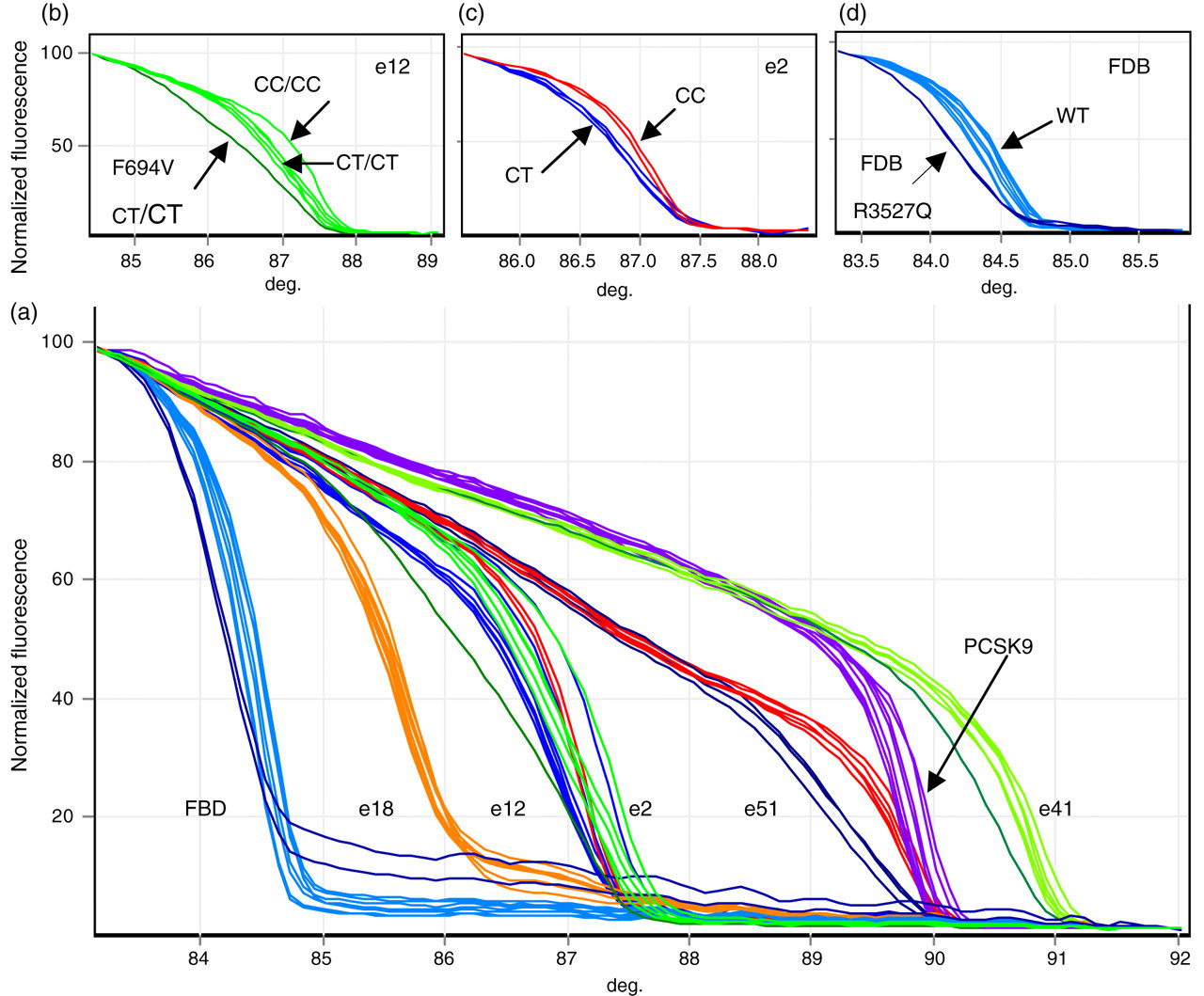
HRM for eight FH samples from Greece, All-Exons Run I. (a) Exons 2, 12, 14, 15, 18 of the LDLR, exon 7 of PCSK9 covering p.D374, and part of exon 26 of APOB covering p.R3527 are run simultaneously for HRM. Exon 14 shows a mutation in dark green (p.F694V). Exons 15, 2 and 12 show polymorphism profiles. The exons 2 and 12 melts overlap and are shown separately in (b) and (c). (b) Exon 12 showing genotypes and the variant p.V613I (note that it has the CT/CT genotype). (c) Exon 2 showing the common homozygote and heterozygote for SNP. (d) Analyses of the APOB fragment, plus profile for two mutation-positive samples
PCR and HRM conditions for the combined fragment Runs I to III and Run IV
HRM, high-resolution melt; PCR, polymerase chain reaction
Runs I to III: both PCR and HRM are performed in the rotor-gene. Eight samples are run for each exon.
Run IV: PCR performed in the GS4 thermal cycler. HRM performed on the rotor-gene. Samples for this exon are run in a single exon run for HRM up to 72 samples, a full rotor
DNA from 28 subjects with FH from Greece was screened for variations in this way and their characteristics are shown in Table 3. Six of the patients had tendon xanthoma and thus a diagnosis of Definite FH (DFH) and the remainder had a diagnosis of possible FH (PFH). The characteristics of the two groups were similar, except that a higher proportion of the DFH group had evidence of CHD. Analysis was carried out using the optimized conditions for the promoter and exons of the LDLR gene together with p.R3527Q in APOB and exon 7 of PCSK9. Samples with variations from the wild-type melt profile in the HRM were identified and sequenced. Overall, 17 of the patients (60%) had a detectable mutation in LDLR; none carried the p.Q3527 mutation in the APOB gene, or the p.Y374 mutation in PCSK9. Eight different variations were found: c.41delT (1 subject), p.W165X (1 subject), p.C173R (3 subjects), p.S286R (2 subjects), p.V429M (4 subjects), p.G549D (4 subjects), p.V613I (1 subject) and p.F694V (1 subject). The individual lipid levels and characteristics of the 17 subjects are presented in Table 4.
Baseline characteristics of Greek FH patients with and without documented tendon xanthoma (TX)
P values are from Fisher's exact test or Mann-Whitney U test due to small numbers
N, number of subjects, HYT, hypertension; LDL-C, low-density lipoprotein-cholesterol; HDL-C, high-density lipoprotein-cholesterol; CHD, coronary heart disease
For traits marked* (values in parentheses) = standard deviations
Mutations detected and characteristics and lipid levels of the Greek patients
CHD, coronary heart disease; T-Chol, total cholesterol; TG, triglycerides; LDL, low-density lipoprotein; HDL, high-density lipoprotein
*TX, tendon xanthoma.
Sözen MM, Whittall R, Oner C, et al. The molecular basis of familial hypercholesterolaemia in Turkish patients. Atherosclerosis 2005;
Lombardi P, Sijbrands EJ, van de Giessen K, et al. Mutations in the low density lipoprotein receptor gene of familial hypercholesterolemic patients detected by denaturing gradient gel electrophoresis and direct sequencing. J Lipid Res 1995;
Hobbs HH, Brown MS, Goldstein JL. Molecular genetics of the LDL receptor gene in familial hypercholesterolemia. Hum Mutat 1992;
Traeger-Synodinos J, Mavroidis N, Kanavakis E, et al. Analysis of low density lipoprotein receptor gene mutations and microsatellite haplotypes in Greek FH heterozygous children: six independent ancestors account for 60% of probands. Hum Genet 1998;
Fouchier SW, Kastelein JJ, Defesche JC. Update of the molecular basis of familial hypercholesterolemia in The Netherlands. Hum Mutat 2005;
Homozygous samples
The obligate heterozygous parents of the Croatian homozygous patient were available for screening, and as shown in Figure 4a they both had a similar variant HRM profile in exon 3 (but not in any other exon) implying they are carrying the same mutation. The child also showed a modest exon 3 variant HRM profile compared with wild type (but not in any other exon), but after mixing PCR product from a sample with no mutation in exon 3 with the homozygote patient to create heteroduplexes, the resulting HRM showed clearly that the homozygote child has the same profile as the heterozygous parents. On sequencing, the parents were found to be heterozygous for the same mutation, p.Q92X, and the child homozygous for this mutation.
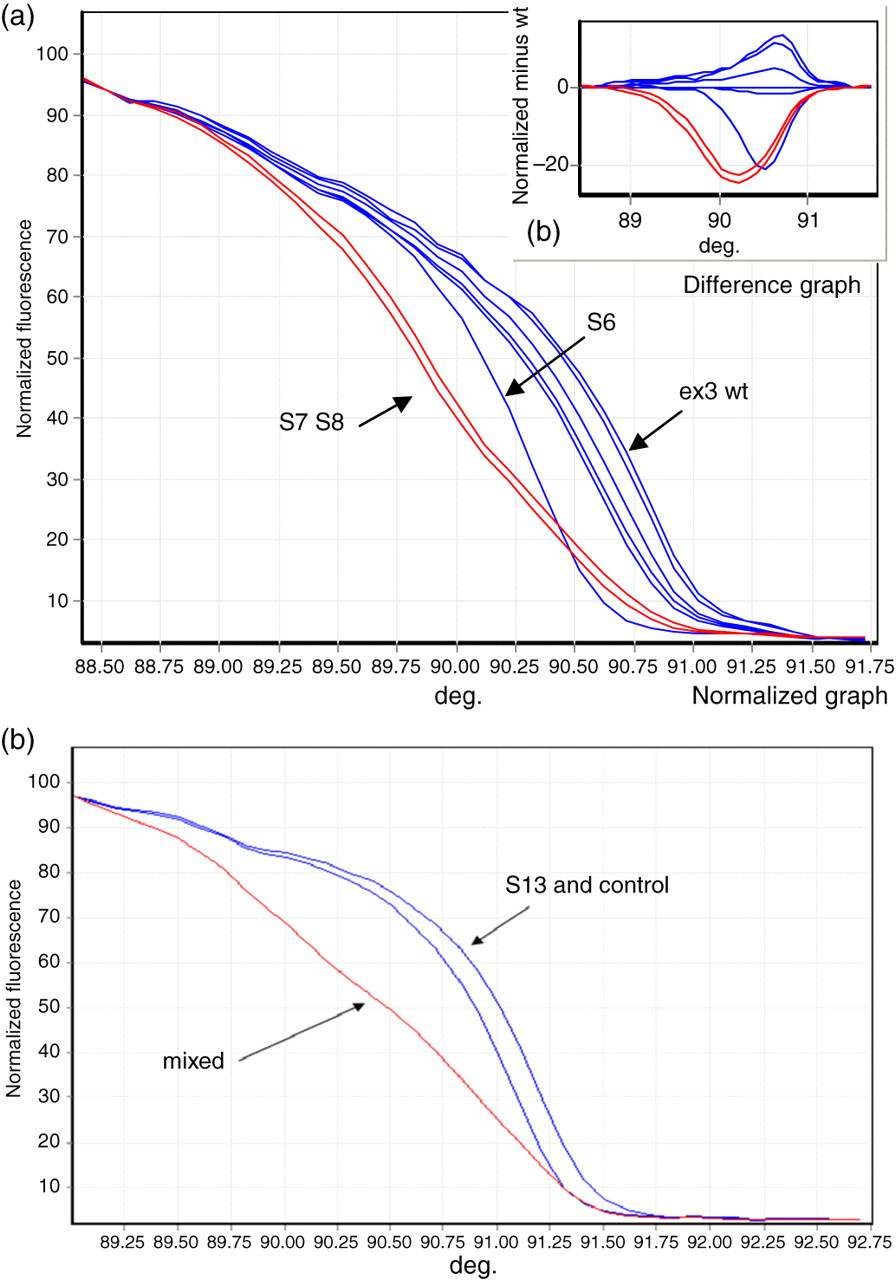
Analysis of DNA from Croatian and UK homozygous FH patients. Figure 4a Exon 3 HRM of homozygous patient (S6) with parents (S7 and S8). (a) The parents (S7 and S8 – red) clearly show the same variant HRM profile implying they are carrying the same mutation. The child S6 also shows a different shift in HRM profile compared with wild type. (b) Difference plot of normalized minus wild type, again showing a clear shift in both parent and child. Figure 4b Showing HRM for exon 10 for the UK homozygous patient (S13) alone and mixed with wild-type DNA. PCR/HRM was performed in the Rotor-gene. Control sample PCR was then mixed with S13 PCR, denatured and cooled before HRM was carried out. The HRM shift in S13 becomes clear when mixed with wild-type exon 10(1) fragment PCR
No relatives were available for the UK homozygous subject, who is of Pakistani origin and whose parents are first cousins. On initial screening no aberrant HRM pattern was found for any fragment. However as shown in Figure 4b, on rescreening all exons mixed with wild-type PCR a variant HRM profile was seen in the exon 10 3′ fragment. The fragment was sequenced and the proband found to be homozygous for p.Y489C.
Discussion
HRM appears to be a sensitive, robust mutation scanning technique that could significantly reduce the time and cost of screening for mutations. The method has been reported to be useful for detecting mutations in the APOB gene in patients with FH 19 and the present report extends this to all three of the known FH-causing genes. Using the Rotor-Gene6000 the HRM method has so far proven to be 100% accurate for 81 known LDLR mutations, and appears to be able to detect all four classes of base changes, which in decreasing order of their occurrence in the LDLR database 4 are A > G/C > T 54.0%, A > C/G > T 26.5%, C > G 12.6% and A > T 6.9%. For the HRM screening the mutations we had available were distributed A > G/C > T 66.7%, A > C/G > T 18.84%, C > G 13.04% and A > T 1.42%. The A > T base change is under-represented overall, and only one example of this class was available and included (and detected) out of the 81 (c.1061A > T, p.D354V). This base change gives the least shift in melting temperature and would therefore be the most difficult to detect using a method based on melting temperature; hence further work is needed to test the sensitivity for detection of this class, which is estimated to make up 7% of single base changes in the human genome. 25 Screening time was rapid. Four PCR–HRM runs can be carried out in one day, equivalent to 288 samples and we have demonstrated that it is feasible to analyse many different exons for small batches of samples within the same HRM run. Both dyes gave melt patterns that distinguished all the known positive mutation controls, although LC-Green gave slightly tighter melt curves in many cases. However, the Syto9 is approximately 20 times cheaper than LC-Green, and this is the dye we now routinely use.
To a large extent, the variant melt pattern seen in a DNA sample heterozygous for a sequence difference is due to the different melting of the heteroduplexes, and thus a DNA sample that is homozygous for a mutation may be more difficult to detect as a distinct shift to the melt profile. Thus the patient homozygous for the mutation in exon 3 did show a distinct shift in the HRM curve, which was flagged for sequencing, while the patient homozygous for the exon 10 mutation did not show a sufficient change in HRM to be recognized as variant. The base change in the exon 3 mutation is CAA > TAA giving rise to a run of three A:T pairings, while the exon 10 mutation is TAC > TGC giving rise to two G:C pairings, which may explain the difference in whether the change in melt pattern is distinct enough to be recognized as variant. However, it is also feasible to screen homozygous patients by mixing DNA with wild-type PCR product for each exon before HRM. This strategy produced a clear result for both homozygous subjects.
Recently the use of HRM for the LDLR has been reported, 20 using the Roche light cycler and LC-Green. While the authors also conclude that HRM is a useful mutation scanning technique, they were unable to obtain adequate melts for the 3′ end of exon 4 and were unable to detect several specific mutations. We also could not obtain adequate melts for the most 3′ region of exon 4 using a three-fragment strategy. Even with a redesign to cover the exon in four fragments the most 3′ fragment could not be optimized on the Rotor-gene using Syto9 or LC-Green and accurate data could only be obtained when the PCR was not performed in the Rotor Gene and DNA binding dye added only after PCR. However, with this approach all exon 4 mutations tested were detected. It is relevant that the problematic region of exon 4 contains an essentially G-rich run (c.694 + 5 to c.694 + 30). Only fragments that do not have this sequence amplify with Syto9 or LC-Green in the PCR mix. Of the examined mutations that were not detected in reference 20 most were not available for us to test, but for example while the authors conclude that c.1706-10G > A may not be detectable since it occurs in the base next to the primer, we were able to detect c.941-39C > T which is also located at the next base to the primer. The authors were not able to detect p.D356N caused by a G > A change (their designation p.D335N), and while we were not able to test this mutation we were able to detect p.D354G (A > G) and p.D354V (A > T). Finally, although the authors conclude that the method is problematic for exons containing common variants, we show that using RFLP analysis (either pre- or post-HRM on the same sample) reduces the difficulties considerably. Sequencing is then required only for those samples where the HRM variation genotype call is not consistent with the RFLP determined genotype, suggesting an underlying mutation.
The p.Q92X mutation detected in the Croatian patient has not been previously reported, but the truncated protein will be non-functional. Although there was no evidence of consanguinity in the family, this child was a true homozygote for the mutation and has a lipid profile expected of a FH homozygous subject. The p.Y489C mutation detected in the UK/Pakistani patient, who as would be expected for a family with consanguinity is truly homozygous, has been previously reported in a patient of Dutch origin 26 but no clinical details were presented. In general, the deletion or addition of a cysteine residue in the LDL-receptor protein has a major impact on its function, since the protein folds incorrectly and is then rapidly degraded intracellularly and does not reach the cell surface. 27 The reported lipid levels in this patient are extremely high, as would be expected for a mutation of such major impact, but it is surprising that the proband currently shows no clinical signs of cardiovascular disease, given her age of 28 y, and that she has also had three uneventful pregnancies. Her parents also show only modest clinical signs of cardiovascular disease, and it would appear that a protective factor or factors are present in this family; whether they are dietary or genetic is unknown (Table 1).
The sensitivity of the HRM method was examined in 28 FH patient samples from Greece. Overall the detection rate was 60% and was 100% in the six ‘definite’ tendon xanthoma-positive patients and 50% in the 22 ‘possible’ FH patients. This is comparable to previously reported detection rates using a combination of methods including SSCP, 8,10,12 dHPLC 11,28 and direct sequencing. 29 It is possible that some of the patients may have a major rearrangement of the LDLR (reported to occur in ∼5% of UK patients) 30 and while this can be detected using methods such as MLPA 31,32 this has not been included here, and nor were the no-mutation samples further sequenced, so we cannot make any statement about the false-negative rate of the HRM method.
In the 28 Greek heterozygous FH patients eight different mutations were detected. Four of these have been reported previously in Greek patients, 33–35 with c.41delT, 36 p.W165X, 37 p.V613I 26 and p.F649V being reported for the first time among FH Greek patients, increasing the total number of FH-causing mutations in Greece to 20. The three commonest mutations in Greece are p.G549D, p.V429M and p.S265R 32–34 and these were detected in four, four and two patients, respectively. One less common mutation (p.C173R, designated Greece-1 27 ) was identified in three subjects. Only p.F694V has not been reported previously in the literature, 4 and the amino acid at this position is phenylalanine in all species except chick, indicating a high degree of species conservation. Using the functional prediction algorithms used in Leigh et al. 4 this variant was predicted to be ‘benign’ by POLYPHEN, but not tolerated by SIFT, and we thus designate this variant as FH causing. However for the pV613I variant, the amino acid at this position is valine only in Human, Macaque, chick and zebrafish, indicating a low degree of species conservation, and this variant was predicted to be ‘benign’ by POLYPHEN, and tolerated by SIFT, and thus the evidence that this variant is FH causing is weaker. However, this variant was the only sequence change identified in this patient, and it has been previously identified in a Dutch FH patient. 26 Whether those with no HRM detected mutation have monogenic hyperlipidaemia due to a non-detected LDLR mutation, or a mutation in a yet to be identified gene, can only be determined by further sequencing or detailed family co-segregation studies. It is also likely that at least a proportion of the subjects does not have true monogenic hyperlipidaemia, but rather have the clinical diagnosis due to a mixture of polygenic lipid raising and environmental influences.
The major limitation for the method is the problem of common SNPs. The LDLR has several exonic and intronic common polymorphisms included in the amplified fragments. Since HRM relies on variations in melting between heteroduplexes and homoduplexes, differences in melting from the wild type are seen with these polymorphisms. These variations in melt can confound mutation detection as a mutation may give a variant melt curve very like the polymorphism. For the single exon runs with 72 subjects this was not a major problem as samples could be genotyped before or after HRM by RFLP analysis and grouped accordingly. It was also feasible to group samples electronically to some extent by inferring genotype, as the profile for each genotype was clearly distinguishable for all exons. However, in the combined fragment protocol, the eight individual HRM profiles were not sufficient to be able to analyse the different polymorphism genotypes separately without RFLP analysis, and in this case any fragments showing a variant pattern, even potential polymorphisms, were sequenced. This limits the effectiveness of the approach.
A second potential limitation of the method would be if false negatives occurred due to the presence of common or rare variants within the sequence of the amplifying primers, which then prevent their annealing and subsequent PCR of this exon. If these occur on the same allele as the mutation only PCR product from the other (wild type) allele would be amplified (or the mutation carrying allele would be poorly amplified) and no melt profile difference would be observed. If the sample was a true homozygote (i.e. also homozygous for the primer SNP) no PCR would occur and this would be seen as a failure to amplify or ‘deletion’. For a compound heterozygous sample the same logic as above would occur for each mutation-containing exon, but the probability that both mutations also have an unreported rare variant in one of the primer regions is very small. To overcome this when primers were designed, regions containing any reported SNPs were avoided using information from the LDLR SNP database. 4 Subjects heterozygous for mutations in each of the exons (apart from 16, 17 and 18) and the promoter were identified with this set of primers indicating that this is not generally a problem.
The 2008 UK NICE guidelines recommend DNA testing for all patients with a clinical diagnosis of FH, 38 and with the population prevalence of 1/500 there are expected to be over 120,000 FH patients in the UK, clustered in roughly 25,000 different families. While the available rapid ARMs kit that tests 20 common mutations is estimated to detect up to 50% of the detectable mutations, 14 this still leaves a large number of subjects where screening of the whole LDLR gene will be required, and the HRM method described here may be of utility.
DECLARATIONS