
Abstract
The primary antibody deficiency syndromes are a group of rare disorders characterized by an inability to produce clinically effective immunoglobulin responses. Some of these disorders result from genetic mutations in genes involved in B cell development, whereas others appear to be complex polygenic disorders. They most commonly present with recurrent infections due to encapsulated bacteria, although in the most common antibody deficiency, Common Variable Immunodeficiency, systemic and organ-specific autoimmunity can be a presenting feature. Diagnostic delay in this group of disorders remains a problem, and the laboratory has a vital role in the detection of abnormalities in immunoglobulin concentration and function. It is critical to distinguish this group of disorders from secondary causes of hypogammaglobulinaemia, in particular lymphoid malignancy, and appropriate laboratory investigations are of critical importance. Treatment of primary antibody deficiencies involves immunoglobulin replacement therapy, either via the intravenous or subcutaneous route. Patients remain at risk of a wide variety of complications, not all linked to diagnostic delay and inadequate therapy. In common variable immunodeficiency (CVID) in particular, patients remain at significantly increased risk of lymphoid malignancy, and regular clinical and laboratory monitoring is required. This review aims to give an overview of these conditions for the general reader, covering pathogenesis, clinical presentation, laboratory investigation, therapy and clinical management.
Introduction
The immunoglobulin repertoire is vital for effective host protection against a wide variety of pathogens. Immunoglobulins are produced by plasma cells, which represent the final differentiation stage of B lymphocytes. B lymphocytes develop from haematopoetic stem cells in the bone marrow and undergo a series of developmental steps during which rearrangement of the B cell receptor genes occurs. Successful expression of first the mu heavy chain and subsequently the light chain (either kappa or lambda) on the surface of the developing B cell allows continued differentiation through the pre-B cell stage to produce a mature, antigen-naïve B cell. This cell then leaves the bone marrow and after receiving an as yet unidentified signal, joins the circulating B cell pool. These events are summarized in Figure 1.
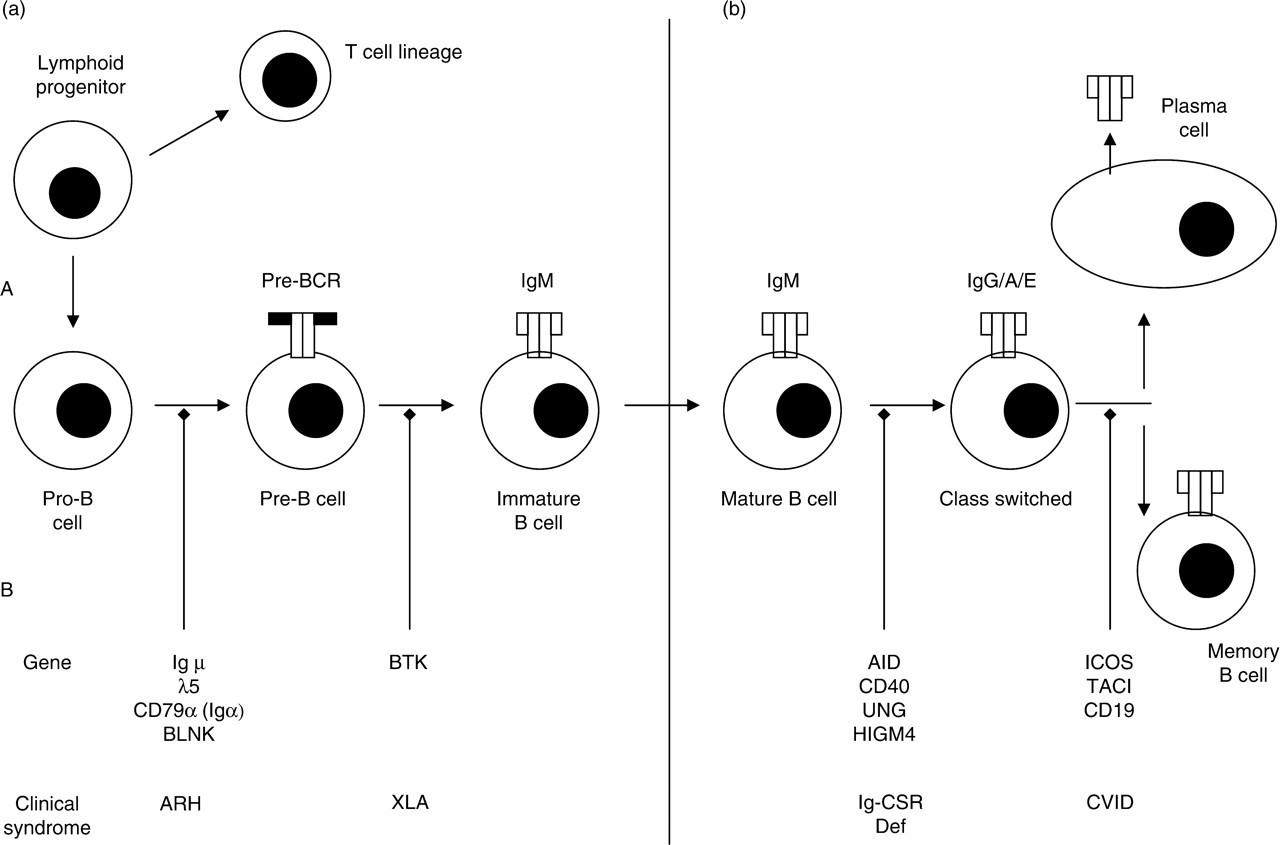
Normal B cell development is shown in the upper half of the figure (a). In the lower half of the figure (b), the genes identified in abnormalities of antibody production and the clinical syndromes they produce are listed at the point in the developmental pathway where the genetic mutation is thought to result in the clinical phenotype. Pre-BCR, pre-B cell receptor (consisting of two immunoglobulin heavy chains and two surrogate light chains); Ig, immunoglobulin; BLNK, B cell linker protein; BTK, Bruton's tyrosine kinase; AID, activation-induced cytidine deaminase; UNG, uracil-N-glycosylase; HIGM4, hyper-IgM syndrome type 4 (unidentified genetic defect); ICOS, inducible co-stimulator; TACI, transmembrane activator and CAML activator; CD19, cluster of differentiation 19; AR, autosomal recessive; ARH, autosomal recessive hypogammaglobulinaemia; XLA, X-linked agammaglobulinaemia; Ig-CSR, immunoglobulin class-switch recombination deficiencies; CVID, common variable immunodeficiency
Encounter with antigen in regional lymph nodes initiates further differentiation of B lymphocytes and leads ultimately to the production of immunoglobulin. Initial immunoglobulin (Ig)M production is replaced by class-switched, high-affinity IgG, IgA or IgE production following interaction between B cells and antigen-specific T cells. IgM plays a critical initial role in binding to intravascular pathogen and in complement activation, while secondary immune responses, characterized by the production of class-switched, high-affinity immunoglobulins such as IgG, IgA and IgE, are necessary for efficient ongoing protection from invading pathogens. IgA fulfils multiple roles in host defence at mucosal surfaces. In the extravascular space IgG is a key effector molecule in the process of opsonization and complement activation. IgE is involved in host defence against parasites. The production of high-affinity IgG against pathogen is a critical component of immunological memory against many infectious organisms, and is a major part of the success of clinically relevant vaccination. The production, regulation and function of immunoglobulins are covered in detail elsewhere, 1,2 but disorders arising as a result of defects in any of the above processes have profound clinical consequences.
Although primary immunodeficiencies are a rare group of diseases, primary antibody deficiencies are by far the commonest of these diseases. In contrast to the majority of primary immunodeficiencies involving other components of the immune system, primary antibody deficiencies can present at any age with peaks in childhood and in the third decade of life. 3,4 Awareness of this information is critical for a prompt diagnosis and initiation of appropriate treatment. It is important to distinguish primary antibody deficiencies from much commoner secondary immunodeficiency states including lymphoid malignancy and HIV infection.
The primary antibody deficiency syndromes are a heterogeneous group of disorders of different aetiologies, in which the final common consequence is an inability to produce effective humoral immunity against invading pathogens. The heterogeneity in pathogenesis of these disorders can produce markedly different clinical consequences, although many similarities between the different syndromes exist. This review aims to provide an overview of these conditions for clinical and laboratory personnel.
Pathogenesis
A number of primary antibody deficiencies arise as a result of single genetic mutations encoding proteins which play a critical role in the development of B lymphocytes.
Early B cell development defects
A number of defects has been described in molecules critically involved in signalling from the pre-B cell receptor. These are autosomal recessive and occur in either sex. 5 Mutations in the mu heavy chain, 6 surrogate light chain lambda5/14.1 7 and more recently cluster of differentiation (CD)79α 8 and B cell linker (BLNK), 9 all produce a similar phenotype with early B cell developmental arrest and hypogammaglobulinaemia (Table 1 and Figure 1).
B cell defects resulting in antibody deficiency
The clinical diseases, peripheral B cell numbers, immunoglobulin patterns, mode of inheritance, genetic inheritance and presumed pathogenesis are listed. See Figure 1 for abbreviations
X-linked agammaglobulinaemia (Bruton's disease)
This is the commonest of the single-gene defects resulting in primary antibody deficiency, with an incidence of approximately 1:380,000, 10 and results from mutations in the gene encoding Bruton's tyrosine kinase (BTK), 11,12 a signalling molecule critically involved in signalling from the B cell receptor. 13 Boys with this condition usually have a profound hypogammaglobulinaemia affecting all classes and absent or extremely low numbers of B cells in the circulation.
Later B cell development defects
B-cell intrinsic immunoglobulin class switch recombination (IG-CSR) deficiencies (previously termed hyper-IgM syndromes)
These are a group of disorders resulting from the failure of effective class-switching processes from IgM to other Ig isotypes, often resulting with an elevated or normal IgM with low IgG and IgA (Table 1 and Figure 1). 14 In contrast to X-linked hyper-IgM syndrome (HIGM), owing to CD40-ligand deficiency, which results in a combined immunodeficiency with opportunistic infection, 15,16 the autosomal recessive forms result in a clinical phenotype similar to other antibody deficiency states. Mutations in genes encoding activation-induced cytidine deaminase (AID), 17 uracil-N-glycosylase (UNG) 18 and at present other unidentified genetic mutations 19 lead to a number of subtypes of Ig-CSR deficiencies, often characterized by normal or elevated IgM levels and enlarged lymph nodes with disordered architecture.
More recently, mutations in other genes encoding proteins involved in the later stages of B-cell development have been identified as potentially causative in primary antibody deficiency states. These include inducible co-stimulatory receptor (ICOS), 20 transmembrane activator and CAML (calcium-modulator and cyclophilin-ligand) interactor (TACI) 21 and CD19. 22 More recent genetic studies 23,24 suggest that only certain TACI variants constitute an increased risk for disease development and the role of these genetic disorders in primary antibody deficiency requires further study.
Complex genetic and environmental disorders
Common variable immunodeficiency
This is the most common of the primary antibody deficiencies with an incidence of between 1/30,000 and 1/50,000. It in itself is a heterogeneous condition with varying phenotypes. Although some of those individuals currently labelled with CVID may subsequently be identified to have single genetic defects as a cause for their clinical phenotype, it is likely that there will remain a large group with a polygenic disorder resulting from genetic predisposition and environmental triggers. Most patients have a significant reduction in at least two of the three immunoglobulin isotypes routinely measured, and variable numbers of circulating B cells, but always in excess of 1% of lymphocytes. At presentation many patients have normal or elevated IgM levels that fall during therapy.
IgA deficiency, the subject of a recent review in this journal, 25 has a degree of overlap with CVID. There is a common restricted human leukocyte antigen (HLA)-extended haplotype, suggesting possible common aetiology between these two conditions. 26,27 Family members of patients with CVID have a higher incidence of IgA deficiency, in addition to autoimmune diseases, the presence of autoantibodies and malignancy. 26
Other disorders of unknown aetiology
A disorder in which individuals fail to mount an effective response to polysaccharide antigen, termed specific antibody deficiency, may also be part of the CVID disease spectrum. 28–30 IgG subclass deficiency, defined as an IgG subclass level that is more than two standard deviations below the normal mean for age, remains on the International Union of Immunological Societies/World Health Organization (IUIS/WHO) list of primary immunodeficiencies, 31 although it is unclear whether isolated IgG subclass deficiency reproducibly results in clinical manifestations in the absence of other immunological defects, e.g. the failure to respond to polysaccharide vaccination. In older individuals, non-malignant tumours of the thymus can produce a syndrome of hypogammaglobulinaemia sometimes accompanied by red cell aplasia (Good's syndrome). 32
Clinical presentation
The IUIS have collated clinical phenotypes for a wide range of primary immunodeficiencies, including the commoner primary antibody deficiencies. 31 Infections with encapsulated bacteria such as Haemophilus influenzae and Streptococcus pneumoniae are the commonest presenting features of the antibody deficiency syndromes. 3,4 Other cohort studies 33–35 have identified respiratory tract infections (including sinusitis), gastrointestinal infections, meningitis, septic arthritis and osteomyelitis as consistent common infectious presenting features in this group of patients. Often, infections respond to standard treatment, only to recur once therapy has been finished. Prolonged and recurrent infection can have significant consequences: in children poor growth and developmental delay, often as a result of end-organ damage in the respiratory tract can occur. In adults, in addition to the debilitating effects of recurrent infection, end-organ damage, particularly of the respiratory tract, remains a significant problem. Bronchiectasis and chronic sinusitis are common complications before the diagnosis is made and appropriate treatment initiated. 36–38 The presence of structural damage frequently results in colonization and subsequent infection by other bacterial pathogens, such as Staphylococcus aureus and Pseudomonas species. It is, at present, unclear as to whether patients with primary antibody deficiency syndromes are more susceptible to infection with these particular organisms. Although bacterial infections are the most common, patients with the CVID spectrum of disorders, are prone to fungal, viral and protozoal infection, including those with opportunistic organisms. This may be related to the presence of T-lymphopenia or evidence of T-cell dysfunction.
In addition to these infective presentations, dysregulation of the immune system is often seen in CVID. This is illustrated by the observation that patients can present with systemic or organ-specific autoimmunity. 3,4,39 This is most commonly haematological, e.g. autoimmune haemolytic anaemia. Systemic autoimmunity including lupus-like conditions and rheumatoid arthritis appears to be less common. Other organ-specific autoimmunity, e.g. pernicious anaemia secondary to autoantibodies directed against intrinsic factor is also common and can be the presenting feature of the condition.
A subgroup of patients with CVID can present with, or develop during the course of their disease, a granulomatous syndrome affecting the liver, spleen, lungs and gastrointestinal tract. 3 These patients can often appear similar to patients with other granulomatous conditions such as Crohn's disease or sarcoidosis and this can lead to diagnostic confusion and delay in appropriate therapy.
Laboratory investigation
Initial investigations
Initial assessment of possible antibody deficiency should focus on establishing the nature of the antibody deficiency state. Nephelometric methods for serum immunoglobulin estimation are highly reliable and reproducible in the presence of variation in polyclonal immunoglobulin concentrations. 40 Significantly reduced immunoglobulins may indicate a primary lack of production or secondary immune paresis or, more unusually, protein loss. In the latter case, a relative reduction in IgG relative to IgA and IgM can often be seen, usually accompanied by reduced serum albumin. Performance of current nephelometric and turbidimetric assays for the measurement of serum immunoglobulins in external quality assurance schemes is generally excellent although there are no data on the sensitivity and specificity of these assays relating to primary antibody deficiency in unselected populations.
At presentation, serum protein electrophoresis should be performed in all adult cases. This is critically important to exclude a secondary antibody deficiency state due to a lymphoid malignancy with a serum paraprotein, which is far more common than a primary immunoglobulin deficiency state. When a monoclonal band is detected, immunofixation should be performed to type the paraprotein (gamma, alpha and mu heavy chains; kappa and lambda light chains). Electrophoresis and, if necessary, immunofixation of concentrated urine for the presence of monoclonal free light chain is an additional important initial investigation.
Follow-up investigations
Serum immunoglobulins, along with serum and urine electrophoresis, should be regarded as first-line investigations. However, it is important to appreciate that normal serum immunoglobulins, in the absence of any paraprotein, do not exclude a diagnosis of primary antibody deficiency. IgG subclass deficiency, characterized by a reduction greater than two standard deviations below the reference range for any of the four IgG subclasses, is a recognized antibody deficiency, 31 although it is unclear whether isolated subclass deficiency provides an adequate explanation for the clinical status of an individual experiencing recurrent infection. There should be a low threshold for the addition of IgG subclass assays to screening requests in the presence of appropriate clinical details.
Such individuals may also have a relative inability to respond to capsular polysaccharide antigens, and deficiencies of antibody levels to capsular polysaccharides may identify an additional or overlapping group of individuals at risk of recurrent infection. Measurement of specific IgG to tetanus toxoid,
41
the polyribophosphate of H. influenzae type B,
42
and to specific polysaccharides from S. pneumoniae,
43
all now performed using enzyme immunoassay, can be a helpful aid to the diagnosis of either isolated specific antibody deficiency or in the assessment of an individual with moderately reduced immunoglobulin concentrations.
31
Many laboratories at present use assays developed in-house to measure these specific immunoglobulins, although commercial kits are available. There is a considerable intra and interassay variation and the overall performance of these assays in external quality assurance schemes is poor (UK NEQAS Annual Report 2007;
Following the identification of a primary defect in immunoglobulin production, lymphocyte surface marker analysis, undertaken by flow cytometry using a whole blood methodology, should be used to measure the percentage and absolute numbers of T-cell subpopulations (CD4 and CD8 T-cells), B and natural killer (NK) cell numbers. These investigations are critical in identifying boys, and exceptionally male adults, with X-linked agammaglobulinaemia (XLA), and children of either sex with early B-cell developmental defects. In adults, the identification of B-lymphopenia should trigger a radiological assessment for the presence of a thymoma. Reversal of the CD4:CD8 ratio often with a degree of CD4 lymphopenia, is seen in around 20% of cases of CVID. 4 It has been known for a considerable time that individuals with this condition can have a varying degree of T-cell dysfunction and markedly reduced CD4 cell numbers may identify a subgroup at greater risk of opportunistic viral and fungal infection. Functional T-cell responses should be assessed using mitogens such as phytohaemagglutinin. The uptake of tritiated thymidine with subsequent beta emission enumeration remains a standard method for the evaluation of proliferative responses. There is evidence that poor phytohaemagglutinin responses may be associated with a worse prognosis and life-expectancy. 4
More recent work has attempted to identify subgroups of patients with CVID on the basis of maturation and sustainability of the memory B-cell compartment. Memory B cells, defined by the presence of surface CD27, can be divided into those which have not undergone class-switching (IgM+) and those that have (IgM−). A number of studies have used broadly similar approaches to identify subgroups of CVID patients within those with normal total B cell numbers – those with no memory B cells, those with no class-switched memory B cells and those with a normal memory B cell compartment. 44–46 There is evidence to suggest that this heterogeneity in B-cell phenotype may predict complications, in particular granulomatous and autoimmune disease, 47 although this remains to be fully elucidated. However, those with low or absence of memory B cell populations may have a worse clinical outcome. 48
Other investigations following diagnosis
In addition to the immunological investigations outlined above, a number of additional investigations should be performed once the diagnosis is established. These should include a full blood count looking for signs of cytopenia, iron, B12 or folate deficiency or an increased reticulocyte count indicating haemolysis. In addition, an assessment of renal function should be undertaken. Liver function is of critical importance as patients may develop both infectious and granulomatous complications of their disease. Baseline assessment is therefore vital. Selected patients may require an assessment of the absorptive function of the gastrointestinal tract.
Although patients with common variable deficiency fail to mount an appropriate antibody response to pathogen, they may paradoxically produce auto-antibodies particularly directed against haematological system components, the endocrine system and the gastrointestinal tract. These should be screened for clinically appropriate cases. Baseline assessment of pulmonary function, including high-resolution computed tomography (CT) of the chest and pulmonary function tests, should be undertaken pretreatment. In addition, any sputum produced should be characterized for pathogen content.
Clinical management
Once a diagnosis has been made this should be fully explained to the patient and their family. The implications of this lifelong diagnosis and potential complications should be outlined at an early stage. Therapeutic options should then be discussed with the patient and appropriate written information given to allow them to make an informed choice.
Immunoglobulin replacement therapy
For the majority of patients, treatment will focus on the replacement of immunoglobulin which can be given by the intravenous (i.v.) or subcutaneous (s.c.) route. Plasma from donors is subjected to a number of premanufacture safety checks, including donor selection, to optimize safety against a number of plasma-borne viruses including HIV, hepatitis B virus (HBV), hepatitis C virus (HCV), parvovirus B19 and West Nile Virus. Unfractionated plasma is frozen to atleast −20°C within 72 h of collection, before a period of quarantine. Fractionation of plasma to selectively produce IgG is complemented by chromatography to increase IgG recovery, following which a number of additional viral inactivation steps, including solvent–detergent treatment, caprylate treatment and nanofiltration, are employed to minimize viral transmission risk. 49 Current methods of viral inactivation and removal do not necessarily neutralize prion agents, but experimental spiking studies suggest that modern processes ensure significant removal of prions. 50 The end product of the process is solubilized and stabilized at various concentrations before being made available for either i.v. or s.c. use. The development of more stable i.v. immunoglobulin preparations in the late 1970s established the i.v. route as the route of choice and this has remained the case in the USA and in most of Europe. In northern Europe and the UK there has been a continued interest in and use of s.c. immunoglobulin as an alternative route for replacement therapy.
Efficacy of immunoglobulin replacement therapy
There are no direct comparative data looking at the distribution of immunoglobulin following i.v. or s.c. routes. High-dose i.v. immunoglobulin may produce FcReceptor saturation, and since metabolism of IgG follows first-order kinetics, higher doses of IgG given intravenously may not produce significant increases in tissue distribution. Although there is little physiological data on the tissue distribution following s.c. immunoglobulin infusion, pharmacokinetic data indicates a ‘smoother’ profile of IgG. 51,52 The UK–Swedish study using a 2-yr timeframe with a crossover design and infection as the primary endpoint revealed no significant differences in efficacy or tolerability between IVIg as against SCIg. 53
There are no randomized controlled trials comparing immunoglobulin with no treatment or placebo. However, i.m. immunoglobulin used in the Medical Research Council trial in the UK in 1950s and 1960s, results in such low plasma concentrations of immunoglobulin that it is now commonly used as a surrogate marker of no therapy. The use of IVIg has contributed to a significantly improved survival rate in primary antibody deficiencies. 54 There are numerous cohort studies demonstrating that immunoglobulin therapy reduces the rate of bacterial infections, days of antibiotic usage, days of fever and hospital admissions. 55–57 Diagnostic delay has been shown to be associated with higher morbidity measured by infection scores. 54 Long diagnostic delay is associated significantly with subsequent reduced lung function. 58
Risks of immunoglobulin therapy
Risks from immunoglobulin are divided into two types: infusion-related adverse events and the risks of transmission of blood-borne viruses by the immunoglobulin product. Infusion-related risks of adverse reaction have been reduced considerably in recent years, owing to improved manufacturing processes (see above), as demonstrated in several trials of new i.v. products. 59–61 Viral transmission has not been reported since the last outbreak of HCV in 1994. 62,63
Home therapy
Patients and their carers can be trained to undertake therapy at home. 64,65 Once patients are stabilized and can tolerate therapy, usually with i.v. immunoglobulin, they should be offered the option of home therapy. Many UK centres have experience and expertise in training patients to self-administer i.v. immunoglobulin at home.
Complications of primary antibody deficiency and their management
Patients with primary antibody deficiencies remain susceptible to a variety of infective complications (Table 2). Many patients still experience diagnostic delay. 35,66 The consequence of this may well be structural lung damage or other end-organ complications. While immunoglobulin given by either route is highly effective in protecting patients from invasive bacterial infection, patients continue to experience breakthrough infection; particularly those with end-organ damage. Persistent infection may be a problem. Antibiotic prophylaxis, either continuous or intermittent, is often used in these circumstances, although there is no clear evidence for its efficacy.
Common infectious complications of antibody deficiency
The common clinical presentations and causative organisms are presented. XLA, X-linked agammaglobulinaemia
Patients with antibody deficiency syndromes can develop a wide variety of end-organ complications – these are summarized in Table 3.
Summary of organ-specific complications
The organ systems and clinical complications of primary antibody deficiencies affecting them are listed
Respiratory
End-stage lung disease with the development of cor pulmonale and respiratory insufficiency is the commonest cause of morbidity in primary antibody deficiency. 3,4 The commonest pathogens isolated from the sputum of patients are H. influenzae, S. pneumoniae and S. pyogenes with Pseudomonas aeruginosa and Moxarella catarrhalis occurring less frequently. 67–70
The precise prevalence of bronchiectasis in patients with primary antibody deficiency is unknown. Cohort studies have reported prevalence rates ranging between 17% and 76%. 4,38,67,71–73 Other pathologies including pyogenic lung abscess, empyema and cryptogenic organizing pneumonia have been described. 56,69 As indicated in the previous section, higher dosing with immunoglobulin replacement therapy may be of benefit to individuals with structural lung damage, but physiotherapy and antibiotic regimens have a role. There are no data to indicate whether continuous rotational antibiotic therapy or intermittent single-agent therapy are efficacious, although both are widely used.
Granulomatous lung disease is more commonly associated with dyspnoea, splenomegaly and restrictive lung defect and is associated with a worse prognosis. Ground glass appearances on chest radiograph and CT scan, often with a restrictive pattern of abnormality on pulmonary function testing, suggest the diagnosis. 74,75 High-dose steroids have been used for therapy, based on their benefit in other interstitial lung diseases. More aggressive immunosuppression may be required, despite the patient's underlying immunodeficiency state.
Gastroenterological
Gastrointestinal disorders are reported in up to nearly 50% of patients with primary antibody deficiency. 33,76 Chronic diarrhoea of unspecified cause occurs commonly in both CVID and XLA. 3 Infectious diarrhoea is relatively common – the most frequently identified organisms are Giardia, Campylobacter spp. and Salmonella spp., all of which occur with increased frequency in untreated patients with both CVID and XLA. Infections require prompt and prolonged high-dose antibiotic therapy – subclinical infection may be common. Small bowel villous atrophy can be found in those patients investigated by endoscopy, occurring at an overall frequency of around 2.5% in cohort studies, 3,4 and possibly higher in children. 68 Some of these features are similar to gluten-associated enteropathy, but may not respond to gluten withdrawal. Atrophic gastritis is reported in patients with CVID, but not XLA, and in some cases is associated with pernicious anaemia. Hyperplasia of gut lymphoid tissue is seen frequently in patients with CVID. Inflammation of both small and large bowel is also reported. Various types occur, including autoimmune, Crohn's-like and non-specific. 77,78 Oral steroids have been used for therapy of various inflammatory complications, based on anecdotal evidence.
Hepatic
HCV infection has been associated with infusion of contaminated batches of immunoglobulin in the past (see above). Other forms of infectious hepatitis can occur, although this is not a frequent complication. 3 Abnormal liver function is common, and a clear cause may not be definable. 4 Hepatomegaly may be caused by granulomatous infiltration, and is often associated with splenomegaly. Granulomatous liver disease may regress with instigation of immunoglobulin replacement therapy. More aggressive disease may require immunosuppression.
Haematological
A variety of haematological disorders are recognized in association with primary antibody deficiency. Most frequent are autoimmune cytopenias, which may occur as the initial presenting feature of immunodeficiency. In the largest case series of CVID reported (326 patients), 11% had a history of autoimmune haematological disease. 79 Broadly comparable numbers of patients with immune thrombocytopenia (ITP), autoimmune haemolytic anaemia (AIHA) and Evans' syndrome (both ITP and AIHA) were reported. The largest UK case series 3 reported haematological complications overall in 39% of patients with CVID, and 18% with XLA. Lymphopenia was included and was common. Five percent of patients with CVID had AIHA, and in half of these the AIHA occurred before the diagnosis of immunodeficiency. Neutropenia was seen in 21 patients, the majority of whom suffered from XLA. Such complications often respond to conventional management with steroid therapy. Splenectomy is best avoided as this increases infection risk. Rituximab has been used successfully to treat ITP associated with CVID.
Malignancy
The increased risk of malignancy in patients with primary antibody deficiency is well-recognized. 80 Estimates of the overall increased risk vary from 1.8-fold to 13-fold 81–83 and appear to be linked with the primary antibody deficiency rather than the genetic background of the individual. 81 The risk of cancer in patients with CVID varies with age, from 2.5% in patients under 16 years, to 8.5% in the age group of 16 years and above. 84 Lymphoreticular malignancies are most frequent, with non-Hodgkin's lymphoma, largely B cell in origin, predominating. 83,85,86 Epithelial malignancies also occur with increased frequency, particularly gastric carcinoma. 67,82 Although the risk is particularly high in CVID there is also a significant increase in other forms of primary antibody deficiency. 87
Many case series include patients with unexplained lymphadenopathy and/or splenomegaly. Some of these have granulomatous changes, and some have ill-defined lymphoproliferative disorders, both of which can also occur and coexist in other organs. Epstein-Barr virus is found in association in only a minority of patients with lymphoproliferative disease. Monoclonal or oligoclonal populations of lymphocytes may be found in lymphoproliferation and lymphoma, and the distinction between these is difficult in patients with primary antibody deficiency. 86
Neurological
Central nervous system infection can be both a presenting feature and complication of primary antibody deficiencies. One study 3 reported that the causative organisms included S. pneumoniae, Neisseria meningitidis, S. aureus, Escherichia coli, H. influenzae and Listeria monocytogenes, with pneumococcal disease the most common. There is a well-recognized occurrence of enteroviral infection, particularly in XLA. 88 Unexplained neurodegeneration can also occur. 89,90
Rheumatological
Rheumatological complications of primary antibody deficiencies include those due to acute or chronic infection that resolve on appropriate antibiotic therapy and are prevented by the institution of immunoglobulin therapy, 91 seronegative arthritis associated with chronic low-grade infection 92 that resolves once adequate immunoglobulin therapy is started, and associated immune-mediated connective tissue diseases. 93,94
Cutaneous
The different dermatological manifestations of primary antibody deficiency include infections of the skin, eczema-related disorders, erythroderma, vasculitis and autoimmune diseases including granuloma formation, and skin problems characteristic of specific immunodeficiency syndromes. Skin infections may be fungal, bacterial or viral. In the largest case series of CVID, there were a small number of patients who presented with associated infections (e.g. herpes zoster), urticaria/angioedema and alopecia. 4 Children may have coexistent atopic disease, but there does not appear to be an increased prevalence in primary antibody deficiency. 95
Clinical monitoring
Patients with primary antibody deficiencies require regular clinical monitoring to assess benefits of therapy and to check for the development of complications and medical comorbidities. Ideally, this should be delivered in specialist centres with appropriate experience in the diagnosis and management of these disorders. Pulmonary function testing and high-resolution CT scanning can be used to monitor for the development of respiratory complications, although silent progression of pulmonary disease can occur. Screening for the development of other end-organ complications (see above) should be undertaken as appropriate. The development of lymphoid malignancy remains a particular concern, and patients should be examined regularly with appropriate questions concerning night sweats, weight loss and other symptoms suggestive of lymphoma. Assessment of the tolerability of immunoglobulin infusions is also required, particularly for patients undertaking treatment at home. Many patients may require a multidisciplinary approach to care, preferably within joint outpatient clinics, but where this is not possible, good communication between specialties is essential.
Laboratory monitoring
In addition to those investigations required depending on individual clinical condition, patients on replacement immunoglobulin therapy require a number of laboratory investigations at regular intervals:
Immunoglobulin levels. Those patients on IVIg should have serum immunoglobulins checked at 3-monthly intervals to ensure that the dose of treatment is achieving levels of IgG appropriate to the patient. The sample should be taken prior to the next therapy to identify trough levels. Changes in infusion dose stabilize after around 3 months with i.v. immunoglobulin therapy. For those patients receiving s.c. immunoglobulin therapy, IgG levels stabilize after several months, and should remain at a steady level within the reference range. It is generally assumed that this stable level should be around 7–9 g/L although there is no direct evidence to support this.
Serum electrophoresis. Patients with primary antibody deficiencies remain at risk of the development of lymphoid malignancy. The identification of serum paraproteins is an important part of monitoring and should be undertaken at regular intervals or if the clinical picture changes. Regular screening for the presence of urinary free light chains may also be required and annual testing is appropriate in the majority of patients.
Liver function tests. The risk of both infectious hepatitis and the development of granulomatous liver disease require liver function tests to be undertaken on a regular basis, with most centres undertaking repeated testing on a 3-monthly basis in stable patients.
Tests for malabsorption. Patients with gastrointestinal complications of CVID or other antibody deficiencies may develop malabsorptive states resulting in vitamin D deficiency, calcium deficiency leading to osteopenia, other vitamin deficiencies and iron or B12/folate deficiency. These should be screened for as clinically appropriate.
Conclusions
The primary antibody deficiency syndromes are a heterogeneous group of complex medical conditions, where early diagnosis results in substantial reductions in morbidity and mortality. Early laboratory identification of reduced immunoglobulins is a critical component of early clinical evaluation. The presence of normal serum immunoglobulin levels does not exclude primary antibody deficiency and further investigations are required in appropriate clinical circumstances. The laboratory should be able to undertake specific antibody measurements or be part of a laboratory network that offers these important diagnostic tests.
Early diagnosis reduces morbidity and allows earlier therapy, usually involving replacement immunoglobulin. This therapy has proven benefit in primary antibody deficiencies, but is not without risk and the decision to initiate therapy should be undertaken by clinicians with experience in the diagnosis and management of these conditions. The immunochemistry laboratory has a critical role to play in monitoring, with repeated testing of both serum immunoglobulins and evaluation of the presence of monoclonal proteins being an important part of management.
Footnotes
Acknowledgement
This article was prepared at the invitation of the Clinical Sciences Reviews Committee of the Association for Clinical Biochemistry.