
Abstract
A 47-year-old man presented with abdominal pain, neck stiffness, severe transient hypertension and unusually dark urine. Cerebrospinal fluid investigations and angiography confirmed the diagnosis of a subarachnoid haemorrhage. Porphyrin studies on the patient and his family demonstrated that the family has acute intermittent porphyria. This is the second case report of an acute hepatic porphyria presenting with a subarachnoid haemorrhage. Acute transient hypertension during the attack of porphyria caused the rupture of an intracranial arterial aneurysm.
Introduction
Cardiovascular features of acute attacks of hepatic porphyria include tachycardia and hypertension and are a result of sympathetic overactivity during the attack. No other cardiovascular disease events or features have been associated with acute abnormalities of porphyrin metabolism. We report the case of a patient presenting with a subarachnoid haemorrhage precipitated by transient hypertension occurring during an acute attack of hepatic porphyria.
Case report
A previously well 47-year-old Caucasian man was admitted to hospital following a two-week illness that started with a mild upper respiratory tract infection and generalized myalgia. His symptoms progressed to abdominal pain, anorexia and vomiting and eventually constipation, oliguria and increasing headache. On admission he was confused, had abdominal tenderness, marked neck stiffness, a positive Kernig's sign and a blood pressure of 200/140 mmHg. A lumbar puncture produced uniformly blood-stained CSF with a high red cell count of 1.2 × 109/mm3. His urine was noted to be unusually dark brown in colour becoming almost black on standing. Subsequent urine porphyrin and porphobilinogen screening tests were positive. Porphobilinogen was detected in fresh urine mixed with Ehrlich's reagent to give a red colour. Porphyrins were detected in the same fresh urine using an extracting solvent and demonstrating red fluorescence under ultraviolet light. He developed a euvolaemic hyponatraemia with a serum sodium of 114 mmol/L, consistent with the inappropriate secretion of antidiuretic hormone. Determination of porphobilinogen in urine, by scanning the spectrum of the product formed with Ehrlich's reagent, demonstrated a urine porphobilinogen of 97 μmol/24 hours (reference range 0–18 μmol/24 hours). Quantitative porphyrin determinations, determined by solvent partition methods, 1 demonstrated a urine coproporphyrin of 1040 nmol/24 hours (reference range 0–245 nmol/24 hours), urine uroporphyrin of 2292 nmol/24 hours (reference range 6–43 nmol/24 hours) faecal coproporphyrin of 138 nmol/g dry weight (reference range 0–107 nmol/g dry weight) and faecal protoporphyrin of 352 nmol/g dry weight (reference range 0–135 nmol/g dry weight). Erythrocyte hydroxymethylbilane synthase (PBG deaminase) activity, measured as moles of uroporphyrin formed per millilitre of washed red cells per hour at 37°C, was within the reference range. These findings were consistent with a diagnosis of an acute hepatic porphyria. 2
Carotid angiography demonstrated an aneurysm of the anterior communicating artery. A frontal craniotomy was performed and a clip applied to the aneurysm. He made a complete physical recovery and his blood pressure fell to 130/80 mmHg over 20 days without antihypertensive therapy. Six months before this admission he had remained normotensive before and after drainage of a perianal abscess under barbiturate-induced anaesthesia.
After discharge from the hospital he suffered major behavioural and personality changes. He was disoriented in time and place, had poor concentration, marked short-term memory loss, loss of emotional inhibition, was sometimes jealous and bad-tempered, and suffered visual and aural hallucinations. He was unable to return to work and died after a further cerebrovascular accident three years later.
His large family was investigated extensively for porphyria (Figure 1). None of the other family members had symptoms suggesting porphyria. Enzyme studies and molecular genetic analysis demonstrated that the family has acute intermittent porphyria. In the next generation, the inheritance was traced by following the mutation in the hydroxymethylbilane synthase gene. Affected family members carry the R173W mutation in this enzyme gene. This mutation is a C-T substitution in nucleotide 517 in exon 10. A highly conserved arginine was converted to tryptophan in the mutant protein. The R173W mutation, one of the more common nucleotide substitution mutations affecting the hydroxymethylbilane synthase gene, is present in about 10% of patients with acute intermittent porphyria in the UK. 3
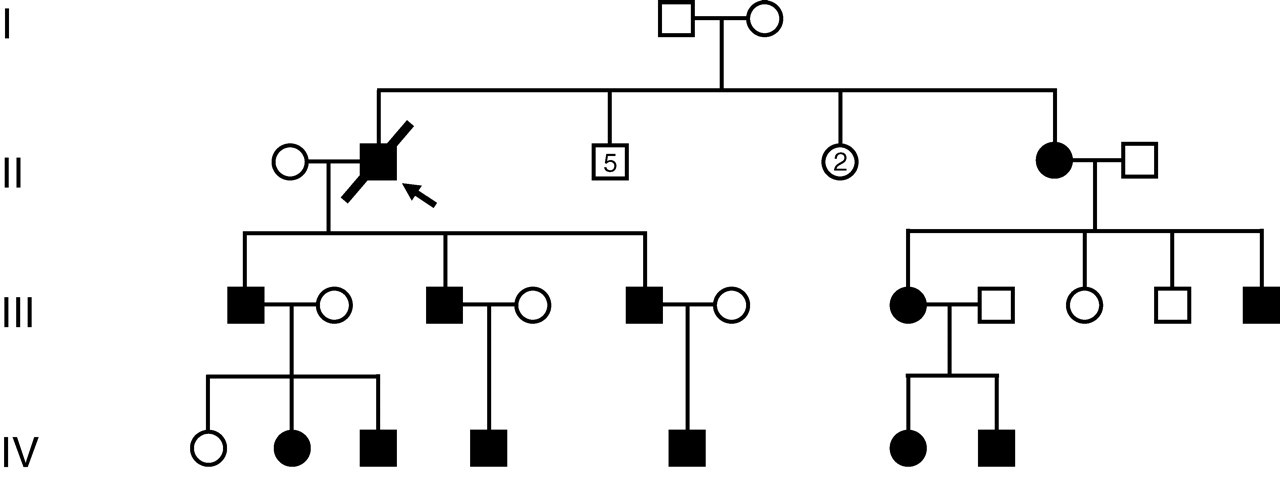
Pedigree showing autosomal dominant transmission of acute intermittent porphyria due to a R173W gene mutation. In the second generation, the deceased proband had five unaffected brothers, two unaffected sisters and one affected sister
Comment
Although the final diagnosis is clear from the genetic analysis, the limitations and pitfalls in the original biochemical methods are recognized and these methods have been superseded by reversed-phase high-performance liquid chromatography (HPLC), the current method of choice for separating all porphyrins of clinical interest. Plasma scanning for porphyrins is now also regarded as a key element in performing a biochemical diagnosis.
An extensive review of the literature does not reveal any association between subarachnoid haemorrhage and acute porphyria 2,4–6 except for one report of a 58-year-old female who was found to have acute intermittent porphyria, a recent subarachnoid haemorrhage and an intracranial aneurysm. 7 The problems posed by the acute intermittent porphyria in the neurosurgical management are discussed by the authors of this report.
A Swedish study of patients with porphyria cutanea tarda, a non-acute porphyria, found a high incidence of stroke on follow-up over a mean of ten years. This association may be due to common risk factors for both porphyria cutanea tarda and cardiovascular disease such as oestrogen therapy, alcohol abuse, diabetes and impaired glucose tolerance. 8 They identified alcohol abuse in 38% of male patients, oestrogen treatment in 55% of female patients, diabetes in 17% or impaired glucose tolerance in 45% of patients tested.
Hypertension is a well-recognized feature of acute porphyria, occurring in 64% of such attacks. 2
The hypertension and tachycardia during the acute attack are thought to be secondary to sympathetic hyperactivity, stimulated by extreme pain, leading to increased production and release of catecholamines. 9 Three-quarters of spontaneous subarachnoid bleeds are due to a ruptured aneurysm, 20% have no identifiable cause and the rest are caused by a variety of rare disorders such as arteriovenous malformations of the brain or spine, arterial dissection, sympathomimetic drugs, tumours or vasculitis. 5 Porphyria has not been recognized as a rare cause.
In our patient, it is probable that an acute attack of acute intermittent porphyria, precipitated by a flu-like illness, produced transient severe hypertension which caused the rupture of a congenital intracranial arterial aneurysm. Initial investigations for a patient presenting with subarachnoid haemorrhage need not include a urine porphyrin screen unless other features suggest porphyria, such as the urine turning dark on standing in the light, especially in populations with a high prevalence of inherited acute porphyria.
Some further educational points illustrated by this case report are:
The importance of investigating atypical findings (dark urine) and not assuming that all features are caused by the presenting disease (subarachnoid haemorrhage). The response to a porphyrinogenic drug, barbiturates in this patient, is unpredictable in a patient with acute porphyria. The initial uneventful exposure to barbiturates does not imply that a subsequent exposure to the same drug would have been safe. There is a great deal of current interest in trying to understand varying individual susceptibility to porphyrinogenic drugs.
10
We conclude that an important message from this case is the value of investigating atypical findings in a disease presentation rather than assuming a single diagnosis.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Dr Gerald E. Levin at St George's Hospital, London and the late Dr D. Nicholson at King's College Hospital, London for initial biochemical investigations on the index patient and the Cardiff Porphyria Service for genetic mutation analyses.