
Abstract
Abnormalities in the DAX-1 gene (dosage-sensitive sex reversal–adrenal hypoplasia gene on the X chromosome) are a well-recognized cause of congenital adrenal hypoplasia. DAX-1 is expressed in the adrenal cortex, gonads, hypothalamus and anterior pituitary, which gives rise to the clinical features of this deletion. Presentations are varied but salt-wasting and/or hypoglycaemia are the most common in an infant, with late onset of hypogonadotrophic hypogonadism. Over 80 different mutations in this gene have been identified. We present three unrelated cases with variable clinical presentations, all with novel mutations in the DAX-1 gene.
Introduction
The condition of congenital adrenal hypoplasia (AHC) was first described in 1948. 1 X-linked cases are due to abnormalities in the DAX-1 gene (dosage sensitive sex reversal–adrenal hypoplasia gene), formally known as NROB1, which is located on the short arm of the X chromosome (Xp21), 2 and encodes an orphan member of the nuclear receptor superfamily. 3 DAX-1 is expressed in the adrenal cortex, gonads, hypothalamus and anterior pituitary. 4 The clinical features include adrenal insufficiency and hypogonadotrophic hypogonadism. Adrenal insufficiency, caused by a failure in the development of adrenal cortex, often presents in early infancy with life-threatening adrenal crisis. Hypogonadotrophic hypogonadism, however usually presents later with pubertal failure. 1–5 AHC has an estimated frequency of 1:12,500 births. 6 It can also present as part of a contiguous gene syndrome together with Duchenne muscular dystrophy (DMD) and/or glycerol kinase deficiency (GKD). 3 To date over 80 mutations in the DAX-1 gene have been identified. 4,7 Here we report three cases, presenting in a variety of ways and all being confirmed by the demonstration of novel mutations in the DAX-1 (NROB1) gene.
Case reports
Case one
A 3700 g male infant was born to non-consanguineous Chinese parents following an uncomplicated pregnancy. He presented at six days of age with jaundice. Physical examination was otherwise unremarkable with no dysmorphic features or hyperpigmentation. Serum thyroid-stimulating hormone (TSH) was elevated at 18 mU/L (reference interval [RI] 0.25–5) and remained elevated at 22 days at 22.6 mU/L with a normal free thyroxine index and normal maternal thyroid antibodies. A presumptive diagnosis of neonatal hypothyroidism was made and he was started on thyroxine replacement (10 μg/kg). This was stopped at five weeks of age following a gradual fall in TSH.
At six weeks of age he presented acutely to hospital following a brief seizure and was noted to be unwell, floppy and lethargic. Initial plasma sodium was 96 mmol/L (RI 136–146 mmol/L) with potassium 7.0 mmol/L (RI 3.5–5.0), glucose 2.5 mmol/L (RI 3.5–7.8) and venous pH 7.39 (RI 7.35–7.45) with a compensated metabolic acidosis. A working diagnosis of acute adrenal crisis was made and he was treated with i.v. hydrocortisone and saline requiring an initial 20 mL/kg bolus to restore circulating volume.
Plasma 17-hydroxyprogesterone was not elevated on the initial neonatal screen and a repeat value was 9 nmol/L (RI 0–15), excluding congenital adrenal hyperplasia due to 21-hydroxlase deficiency. Plasma cortisol at 0700 hours was 178 nmol/L with aldosterone 319 pmol/L (RI 100–800 pmol/L), renin raised to >18.7 nmol/L/hour and plasma adrenocorticotrophic hormone (ACTH) >700 pmol/L (RI 1.8–11.9) confirming the diagnosis of primary adrenal insufficiency. Further studies showed an elevated plasma 11-deoxycortisol of 209 nmol/L (RI 0–30). Plasma triglycerides and creatine kinase were both normal, excluding a diagnosis of GKD and DMD, respectively. Plasma triglycerides were measured by an assay in which they are first converted to free glycerol (Abbott ci8200, Abbott Laboratories, Abbott Park, IL, USA). If GKD was also present then elevated endogenous free glycerol would be detected as artefactually elevated triglycerides. Very long chain fatty acids were not elevated, excluding adreno-leukodystrophy.
Mutation analysis confirmed the diagnosis of AHC. After genomic extraction from whole blood, both coding exons of the DAX-1 gene were amplified using the polymerase chain reaction (PCR) and analysed by automated bidirectional fluorescent DNA sequencing on the ABI 3100 Avant electrophoresis system (Applied Biosystems, Foster City, California). Sequences were compared with the Genebank reference sequence NM_000475. The infant was found to be hemizygous for the nucleotide transversion c.192C > A and also hemizygous for the sequence variant c.498G > A, a previously reported non-pathogenic polymorphism. 7
Currently, aged nine months, he remains well on hydrocortisone and fludrocortisone replacement.
Case two
A male was born healthy at term in 1983. At one month, he presented with a right inguinal hernia and right cryptorchid testis which were corrected surgically, and then subsequently at nine months a left hernia was also corrected surgically.
At 18 months, he presented drowsy following a two-day history of diarrhoea and vomiting. Plasma sodium was 128 mmol/L with potassium 5.5 mmol/L and glucose 2.1 mmol/L. Rotovirus infection was confirmed, fluid rehydration was commenced and he was discharged four days later well with normal electrolytes.
At 24 months he presented again, moribund following a further two-day episode of vomiting. Again plasma sodium was low at 126 mmol/L with potassium 4.7 mmol/L, glucose 1.9 mmol/L and urea 18 mmol/L (RI 1.8–6.4 mmol/L). A random cortisol was 252 nmol/L and synacthen testing confirmed a low cortisol response, 310 rising to 313 nmol/L (RI > 550 nmol/L). 17-Hydroxy-progesterone was normal and plasma renin activity was increased at 20 nmol/L/hour with aldosterone undetectable. Thyroid and adrenal autoantibodies were negative. He was diagnosed with primary adrenal insufficiency with possible associated hypogonadotrophic hypogonadism (due to the previous cryptorchid testis). Treatment was commenced with hydrocortisone and fludrocortisone.
He remained under endocrine review and at age 12 years was noted to have small testes. Bone age was two years delayed. Serum testosterone was low at 0.2 nmol/L, luteinizing hormone (LH) was undetectable at <0.5 IU/L with follicle-stimulating hormone (FSH) at 2.0 IU/L. Human chorionic gonadotrophin (hCG) stimulation (2000 U i.m.) increased plasma testosterone from 0.5 nmol/L to 3.3 nmol/L. Oral testosterone undecanoate 40 mg b.i.d. was commenced for treatment of probable hypogonadotrophic hypogonadism.
At 16.5 years he remained well on replacement testosterone with a normal physique and prepubertal testes. He had normal olfaction.
At this stage, in view of his primary adrenal insufficiency and associated hyponadotrophic hypogonadism, DNA sequencing as described for Case 1 revealed a 51 bp deletion (nt 1068_1118) confirming the diagnosis of AHC.
Case three
A male was born healthy at term in 1970. At one year of age he presented with vomiting and a diagnosis of pyloric stenosis was considered but no lesion was found. Investigations revealed a plasma sodium of 105 mmol/L with a potassium of 7.0 mmol/L. He was discharged following fluid replacement and normalization of his electrolytes.
At four years he presented following a collapse in the context of a vomiting illness. An abnormal response to synacthen and a 10-fold increase in plasma ACTH level confirmed primary adrenal failure. He was commenced on cortisol acetate and fludrocortisone replacement.
He was lost to follow-up for some years, but on review at age 25, appeared clinically hypogonadal with poor musculature, reduced body hair and both testes were small at 8–10 mL. He also had abnormal pigmentation. Sense of smell was normal. Bone mineral density (BMD) was noted to be low with lumbar spine and femoral neck 66 and 78% of normal, respectively.
Further investigations revealed a morning plasma testosterone of 9.2 nmol/L (RI 8–38) with sex-hormone binding globulin 103 nmol/L (RI 10–48) giving a free androgen index of 89 (RI > 400). LH was 1.6 IU/L (RI 2–9) with FSH 4.8 IU/L (RI 2–9).
Hypogonadotrophic hypogonadism was diagnosed and he was commenced on testosterone replacement therapy. DNA sequencing as described for Case 1 revealed an 8 bp deletion (nt 1181_1188).
Now he has normal virilization and sexual function on testosterone, however, his BMD remains low.
Discussion
We have reported three cases of primary adrenal insufficiency secondary to AHC. In each of these cases, the diagnosis of AHC has been confirmed by mutational analysis showing defects in the DAX-1 gene. There are now over 80 mutations reported in the DAX-1 gene 4,7 and the cases above add to these, with all three mutations being novel.
In Case 1, the nucleotide transversion c.192C > A, results in a sequence change from TAC to TAA in codon 64 and a substitution of the normal tyrosine residue with a premature stop codon (Figure 1). This would result in a severely truncated protein which is predictably inactive.
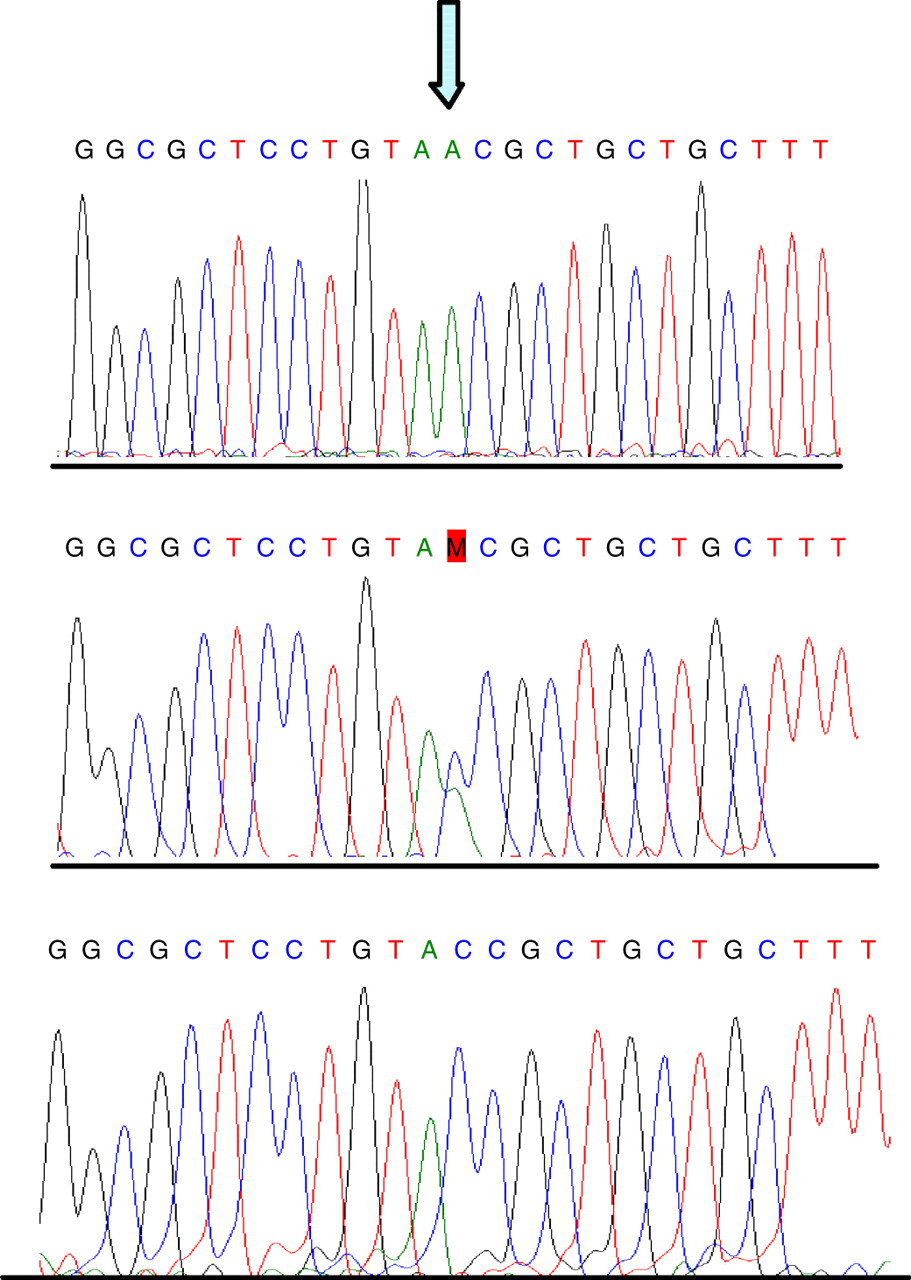
Top panel, showing hemizygous TAA sequence at codon 64 in index case 1 resulting in a premature stop codon, compared with wild-type TAC sequence (bottom panel) coding for normal tyrosine and heterozygous sequence (middle panel) from the carrier mother
With Case 2, the 51 bp deletion (nt 1068_1118) predicts an in-frame deletion of 17 amino acids (residues 279–295) (Figure 2). Few in-frame deletions have been previously described, with only two noted out of all 85 published mutations in 2001. 7
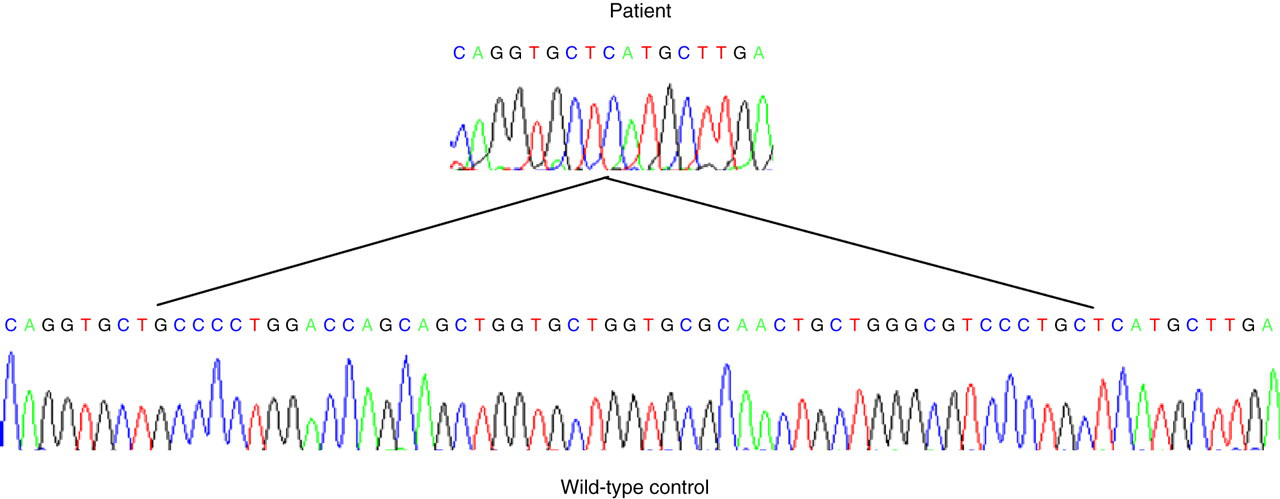
The 51 bp deletion from index Case 2 shown (nt 1068_1118) predicts an in-frame deletion of 17 amino acids (residues 279–295)
The 8 bp deletion (nt 1181_1188) found in Case 3 results in a frame-shift mutation, resulting in a substitution of leucine for methionine at position 316 and the translation of 60 novel amino acids before truncation (Figure 3). Frame-shift mutations appear to be the most common mutations seen in the NROB1 gene. 7
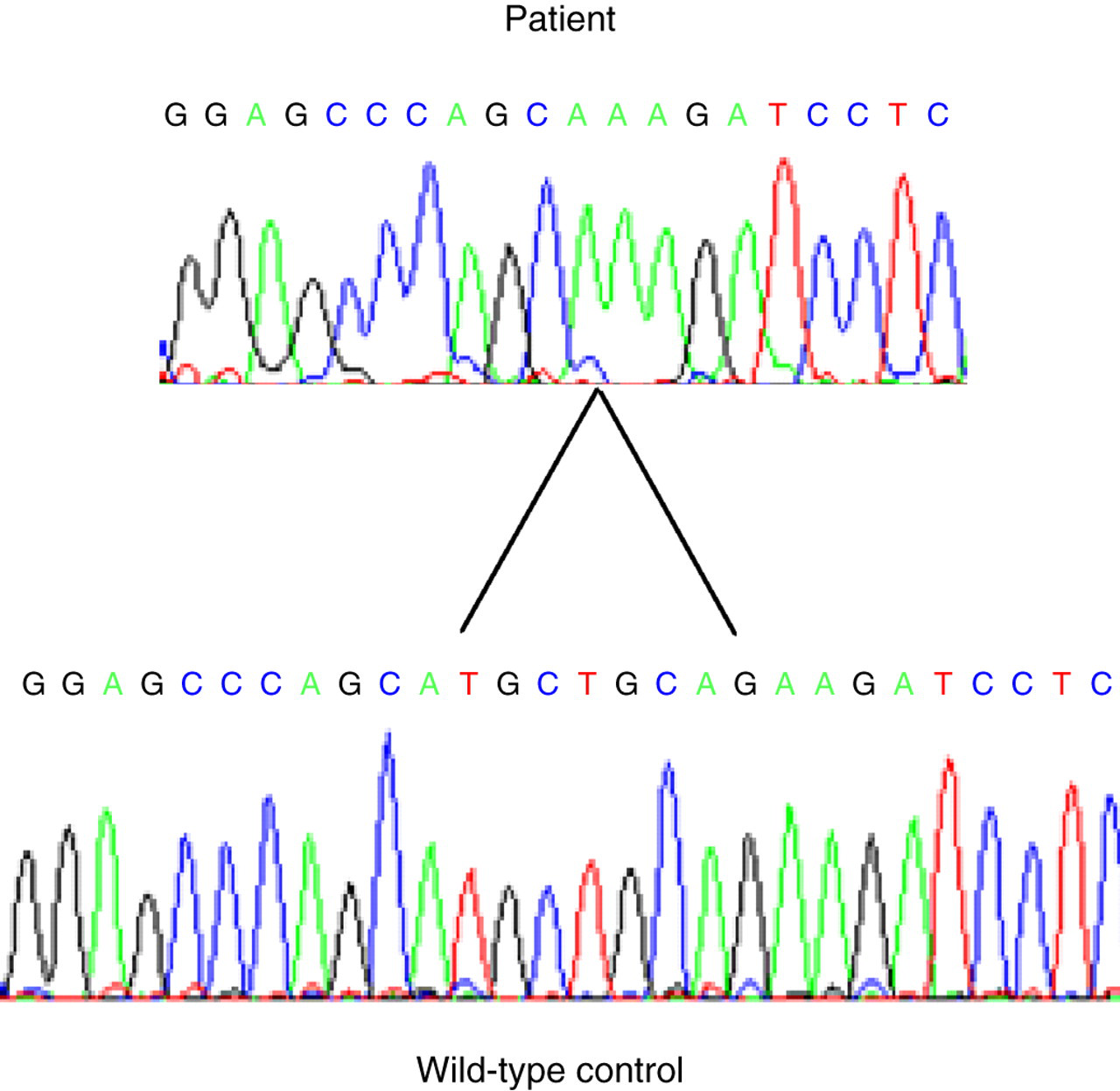
The 8 bp deletion from index Case 3 shown (nt 1181_1188), results in a frame-shift, with a substitution of leucine for methionine at position 316 and the translation of 60 novel amino acids before truncation
In Cases 2 and 3, both mutations occur within a putative ligand-binding domain, LBD2. Surprisingly, the in-frame deletion in Case 2 resulted in a more severe clinical phenotype, with cryptorchidism and earlier adrenal crisis, suggesting that residues between 279 and 295 are critical for ligand-binding. These residues are highly conserved in the LBD of typical nuclear receptors. However, even when individuals with identical mutations in the DAX-1 gene are compared there is a lack of correlation between genotype and phenotype suggesting that additional factors are also important in determining clinical course. 8,9
Clinically, there is a wide variation in the presentation of patients with AHC. 1–5 These three patients nicely demonstrate this phenotypic variation along with the diagnostic difficulties faced by clinicians when faced with AHC. This is evidenced by the delay in diagnosis, particularly in Cases 2 and 3.
All three cases initially presented in the first two years of life. The first case demonstrates a more classic neonatal presentation of AHC with adrenal crisis. Case 1 also presented many years after Cases 2 and 3 and the prompt diagnosis reflects both previous institutional exposure to this condition and the immediate care of a specialist paediatric endocrinology service.
The diagnosis of AHC was not considered until several years after initial presentation in Cases 2 and 3, as awareness of the condition increased with time and diagnostic tools (including mutational analysis) became available. They also showed a more insidious onset of symptoms with multiple presentations, likely in hindsight to be unrecognized adrenal insufficiency. In both cases the development of hypogonadotrophic hypogonadism was the eventual clue to expanding the diagnosis, with AHC later confirmed by genetic analysis.
The elevated 11-deoxycortisol found in Case 1 raised the possibility of 11-beta hydroxylase deficiency, a form of congenital adrenal hyperplasia. This is usually a salt-retaining phenotype, however, and potentially explained in this case by a persistence of a foetal adrenal pattern of steroid synthesis. 10
AHC can also present as part of a contiguous gene syndrome together with DMD and/or GKD. 3 There were no features to suggest the presence of these conditions in the cases described. Furthermore, the demonstration of a point mutation in the DAX-1 gene also helps to exclude a contiguous gene deletion syndrome.
None of the cases identified had a family history of adrenal insufficiency. AHC is an X-linked disorder; therefore identifying a DAX-1 mutation in a patient allows for carrier testing in family members. Screening was conducted in the families of our index cases. The mother of Case 1 was confirmed to be a carrier (Figure 1), as were the mother and sister of Case 2. This allows for accurate genetic counselling and potentially prenatal or even pre-implantation genetic testing.
In conclusion, we have identified three novel mutations in the DAX-1 gene with clinical presentations spread over the past 37 years. The mutations identified include a frame-shift, a nucleotide transversion and a rare in-frame deletion.