
Abstract
Aim
To assess the value of pancreastatin as a predictive factor for identifying patients with neuroendocrine tumours (NETs) who respond poorly to somatostatin analogues.
Methods
A retrospective study of patients with NETs. Patient records from the Northern Ireland Neuroendocrine Tumour Register were interrogated. Those who had pancreastatin concentrations measured on two or more occasions, before and during somatostatin analogue therapy (within the set time-limits) were selected. Data relating to diagnosis, surgery, somatostatin analogue therapy and survival outcome were noted. Data were subjected to univariate and multivariate analysis using Cox proportional hazard model.
Results
Fifty-nine patients with gastroenteropancreatic NETs fulfilled the inclusion criteria. Factors associated with a poor survival outcome on univariate analysis were primary tumour site (P = 0.006) and rapid rise in pancreastatin during somatostatin analogue treatment (P < 0.001). In multivariate analysis, highly significant clinical prognostic indicators were: tumour location (P < 0.001), pre-treatment pancreastatin (P < 0.001) and pancreastatin change (P < 0.001).
Conclusions
This study endorses the finding that pancreastatin is a useful prognostic indicator of neuroendocrine disease. On commencement of treatment, one-third of the subjects showed an immediate negative pancreastatin response to somatostatin analogues, which was associated with poor survival. This is the first study to document such an association. These findings have significant therapeutic consequences. In the presence of a rapidly rising pancreastatin alternative, treatment modalities should be sought.
Introduction
Neuroendocrine tumours (NETs) are a heterogeneous family of uncommon, generally slow-growing tumours derived from enterochromaffin cells. Depending on their primary site, NETs produce peptides and/or biogenic amines. 1 Hypersecretion of these products may result in characteristic NET syndromes. In addition, NETs release a family of proteins known as the granins, the most important of which is chromogranin A (CgA). 2 The granins and particularly CgA are found in increased quantities in endocrine tumour cells. The identification of CgA has been exploited both for diagnostic purposes and to assess response to treatment in patients with NETs. 3 Circulating CgA concentrations are elevated in patients with various peptide-secreting NETs, with the highest concentrations in metastatic serotonin-secreting endocrine tumours of the ileum and colon (midgut carcinoid tumours). Elevated concentrations are also observed in functioning pancreatic tumours and in non-functioning tumours, and other NETs throughout the body. 4,5 As well as being raised in the circulation of patients with NETs, CgA is raised in all conditions where there is an absence or suppression of gastric acid secretion in the stomach. In normal healthy subjects enterochromaffin-like (ECL) cells in the stomach are the main contributors to CgA in the circulation. Where the acid negative feed-back to gastrin is absent, because of autoimmune atrophic gastritis, H2 antagonist or proton-pump inhibitor therapy, gastrin rises and further stimulates the ECL cells to secrete increased concentrations of CgA.
Pancreastatin is a 49 amino acid, post-translational processing product of CgA (CgA 240–288). Before the complete sequence of CgA had been elucidated and CgA whole molecule assays were not available, pancreastatin was used as a surrogate marker for CgA in cells and in the circulation. 6 Circulating pancreastatin concentrations are elevated in patients with NET when the tumour has metastasized to the liver and concentrations are proportional to hepatic tumour burden. 7,8 Some pancreastatin assays that are mid-molecule-specific cross-react strongly with CgA. However, assays that are specific to the N- or C- terminus of pancreastatin measure only the post-translational product of CgA, pancreastatin. Therefore, assays that are specific to pancreastatin may be used for the advantage of patients with NETs. Pancreastatin does not show a rise in patients who are hypo- or achlorhydriac. Pancreastatin assay is used routinely along with whole molecule CgA assay in both the referral laboratories in the UK (SAS Laboratory, Department of Endocrinology and Diabetes, Hammersmith Hospital, DuCane Road, London and Regional Regulatory peptide Laboratory, Kelvin Building, Royal Victoria Hospital, Belfast, UK). In Belfast, pancreastatin assay is used to assess the extent of liver disease in NET patients.
Somatostatin analogues provide symptomatic relief from NET syndromes in more than 60% of patients and form the mainstay of palliative management. Somatostatin analogues act by binding with specific somatostatin receptors (SSTs) located in target tissues. SST receptors are usually found in high density in NETs, which possess up to 100 times more SST receptors than normal tissue. 9 The inhibitory effects of somatostatin, mediated by SSTs, are a result of several mechanisms including: inhibition of adenyl cyclase leading to a reduction of intracellular cyclic AMP concentrations; a reduction of intracellular concentrations of Ca2+ because of decreased influx of Ca2+; and stimulation of tyrosine phosphatase activity. 10 SST analogues may also inhibit cell growth 11 where the antiproliferative action of SST analogues is mediated by the arrest of G1 cell cycle, which activates different signal transduction pathways depending on the receptor subtype.
The hypothesis of this study was that pancreastatin would be a useful predictive factor identifying patients who respond poorly to SST analogues.
Subjects and methods
Approval was obtained for this research from the Research Ethics Committee of Queen's University, Belfast.
Data collection
Any patient diagnosed as having two or more pancreastatin measurements recorded on the NET register at the Regional Regulatory Peptide Laboratory, Belfast and who had been treated with an SST analogue for relief of symptoms, was considered for inclusion. Both pre- and during treatment circulating pancreastatin measurements were required for inclusion and patients were excluded if the time between pre-treatment pancreastatin measurement and treatment commencement exceeded six months. Pancreastatin measurements during treatment were recorded for one year after the commencement of SST analogues. To provide adequate survival data subjects must have commenced SST analogue treatment at least two years before the completion of the study. Patients were followed up till death or till the end date of the study, whichever came first.
Subjects were restricted to those with NETs of the pancreas, foregut (including those of the broncho-pulmonary tract) and ileum or colon. Patients with multiple endocrine neoplasia type 1 (MEN1) or NETs of the appendix were excluded because of the very different disease courses expected in these patients. Only patients with clinical manifestations were included, as all patients were treated with SST analogues for symptom control. All patients had metastatic disease and tumour burden was recorded (number of liver metastases and lymph node metastases). Patients who had previous medical anti-proliferative treatment (including alpha-interferon, radio nucleotide targeted therapy, hepatic artery embolization or liver de-bulking) were excluded. Previous surgical reduction of a NET primary did not exclude patients from the study and this information was recorded. All pathology reports from tumour biopsies were assessed and only those with highly differentiated metastatic endocrine tumours were included. This was recorded where Ki67 had been measured. Ki67 is a measure of proliferative index in tumour cells. Highly differentiated NET cells typically have a very low Ki67, <3%, and this is associated with much better survival than those with Ki67 >3%. All subjects were treated with either Octreotide (Sandostatin® or Sandostatin LAR) or Lanreotide (Somatuline LA® or Somatuline Autogel®). All patients received only one analogue between the pre- and during treatment pancreastatin measurements. Time lapse between diagnosis and treatment with SST analogues were also recorded.
Pancreastatin assay
Circulating pancreastatin was measured in extracts of plasma using an in-house radioimmunoassay. Antibodies were raised in rabbit to synthetic human pancreastatin 39–49 coupled to albumin. Antibodies were specific to the C-terminal region of pancreastatin and cross-reaction with total intact CgA was minimal (<7%). Radiolabelled pancreastatin was prepared using the chloramine-T method. 125I was incorporated into tyr-0 pancreastatin 39–49 (Queen's University, Belfast) and products were purified using high-performance liquid chromatography (HPLC). Standard pancreastatin preparations were prepared from synthetic porcine pancreastatin (33–49) (Bachem UK Ltd, Delphi Court, Sullivan Way, St Helens, UK). Separation of antibody-bound and unbound pancreastatin was made using dextran-coated charcoal. 12 Internal quality control was maintained by including three sample pools with pancreastatin concentrations within the reference range and three pooled patient samples in the pathological range. In addition, there has also been a sample exchange between the Belfast and Hammersmith Laboratories (Proceedings of the UKINETS National Scientific Meeting December 2006). There is no external quality assurance scheme for pancreastatin. Co-efficient of inter-assay variation was quoted as 3.5–13.5% over the range 12–80 pmol/L and intra-assay variation as 8.5–14.0% over the same range. The reference range for pancreastatin assay was 0–25 pmol/L, established in a group of 100 normal healthy subjects (male 50, female 50) in the age range of 20–70 years. This laboratory has shown no change in pancreastatin concentrations with age. Pancreastatin is elevated in patients with NETs who have liver metastases, median 78 pmol/L (range 25–1850 pmol/L, n = 35) and is 12 pmol/L (range 2–22 pmol/L, n = 35) in patients with NETs and lymph node metastases.
Statistical methods
Data were collected on an Excel 2000 spreadsheet and several initial calculations were performed in Excel: age at diagnosis, time between initial pancreastatin measurement and commencement of somatostatin analogue therapy, time between somatostatin analogue therapy and follow-up pancreastatin measurement and survival (measured from the start of therapy). Patients were followed up to a fixed date a minimum of two years post-treatment or death. Data were transferred to SPSS version 11 for Windows (SPSS Inc., Chicago, IL, USA) for further statistical analysis. Univariate survival analysis was performed using Kaplan-Meier survival plots. The following data were analysed: pre-treatment pancreastatin concentration, change in pancreastatin concentration following treatment (during to pre-treatment pancreastatin ratio), age, primary tumour site and surgical resection of the primary tumour. As pancreastatin concentration showed a positive skew, a logarithmic transformation was applied prior to analysis. Comparison of survival between groups was made using a log-rank test. Covariates identified as either having an influence, or being expected to have an influence on survival by univariate analysis was analysed using Cox proportional hazards model. These included age at commencement of treatment, pre-treatment pancreastatin, post- to pre-treatment pancreastatin ratio, site of primary tumour and resection of the primary tumour. P value <0.05 was regarded as significant.
Results
Fifty-nine patients (33 men and 26 women) registered with the Northern Ireland Neuroendocrine Tumour Register fulfilled the criteria and were included in the study. In 57 patients, the diagnosis of NET was confirmed histologically and in two diagnosis was made from positive somatostatin analogue scintigraphy, increased urinary 5-hydroxy indole acetic acid (5HIAA), CgA and neurokinin A with classical carcinoid features of diarrhoea and flushing. All patients who were included had positive somatostatin receptor scintigraphy. All those included had metastatic disease and all histologically proven tumours were highly differentiated endocrine tumours. The characteristics of the study group are recorded in Table 1. All patients were treated with somatostatin analogues for the relief of symptoms. Delay between diagnosis and treatment varied greatly. In 14 of the 29 patients with tumours of the ileum or proximal colon, there was a delay of 1–24 years between tumour resection and commencement of somatostatin analogue therapy during which time the patients were symptom-free.
Characteristics of study group
Median pancreastatin concentration prior to treatment was 90 pmol/L (range 5–8640 pmol/L). In 16 patients (27%), pancreastatin concentration was within reference range before treatment (<25 pmol/L). There was a strong correlation between tumour burden and pre-treatment circulating pancreastatin. Median during-treatment pancreastatin concentration was 75 pmol/L (range 5–13,500 pmol/L). Median survival was 3.7 years from the start of somatostatin therapy.
Of the 59 subjects, 39 (66%) showed a favourable response to somatostatin analogue therapy with respect to circulating pancreastatin concentrations. Median pre-treatment circulating pancreastatin was 130 pmol/L (range 95–8,640 pmol/L). In nine patients (15%), pre-treatment concentrations were within the reference range. Median during-treatment pancreastatin was 35 pmol/L (range 5–3670 pmol/L). Of these, 29 patients had tumours of the ileum or proximal colon, of the lung (4), pancreas (3), gastrinoma (2), gastric (1), thymic (1), mesenteric (1) and of unknown origin (2). Clinical symptoms were initially alleviated in 31 of 39 patients. Median survival was 5.7 years.
In 11 of the 59 subjects (19%), a significant rise in pancreastatin concentration during treatment was observed (between 1.5 and 5 times pre-treatment concentrations). Within this sub-group, median pre-treatment pancreastatin concentration was 50 pmol/L (range 10–920 pmol/L) and three of the 11 (27%) were within the reference range. Median during treatment pancreastatin concentration was 112 pmol/L (range 20–1,620 pmol/L). Eight of these patients had a primary tumour in the ileum or proximal colon and three in the pancreas (one each of insulinoma, Vipoma and glucagonoma). Clinical symptoms were initially alleviated in nine of 11 patients. Median survival was 1.7 years.
Nine patients (15%) who had a tumour of the ileum or proximal colon (7), lung (1) and pancreas (1 gastrinoma) showed a dramatic rise in pancreastatin concentrations following treatment (greater than five-fold increase). Median pre-treatment and during-treatment concentrations were 30 pmol/L (range 10–1190 pmol/L) and 1295 pmol/L (range 145–13,500 pmol/L), respectively. Four (44.4%) of the nine had a pre-treatment measurement within the reference range. Clinical symptoms were initially alleviated in six of nine patients. Median survival in this group was 0.3 years.
Circulating pancreastatin concentrations, pre- and during-treatment, in the three groups described above are shown in Figure 1. Pancreastatin concentrations are shown on a logarithmic scale.
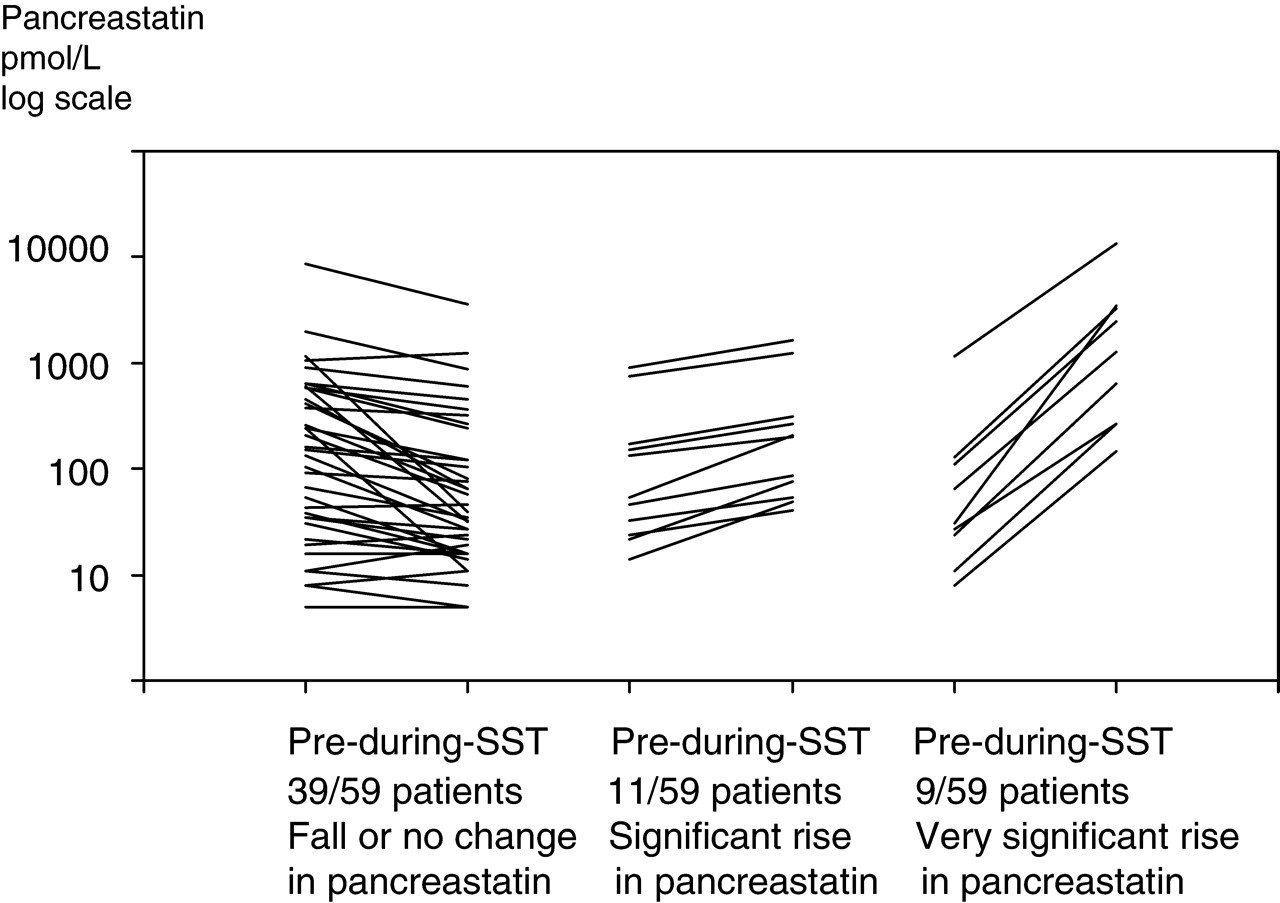
Change in circulating pancreastatin pre- to during-treatment with somatostatin analogues (SST)
Univariate analysis
For initial assessment of the association between pre-treatment pancreastatin concentration and survival, the patients were divided into three groups of approximately equal size; pancreastatin <50, 50–499 or >500 pmol/L. Change in pancreastatin concentration with somatostatin analogue treatment was also divided into three groups: those whose pancreastatin fell or showed no significant change, those with a significant rise but whose during-treatment to pre-treatment ratio was less than five-fold and those with a very significant rise (five-fold or greater). The prognostic value of the following variables was then assessed using log-rank tests supplemented by Kaplan-Meier survival plots: age group, primary tumour location, previous surgical resection of the primary tumour, pre-treatment pancreastatin concentration, ratio of during- to pre-treatment pancreastatin concentration and surgical resection. Type of treatment (Octreotide versus Lanreotide), could not be analysed as there was considerable cross-over of treatment between the during-treatment pancreastatin measurement and eventual outcome.
Pre-treatment pancreastatin concentration did not demonstrate significant differences in outcome (χ 2 = 2.088, P = 0.352). Age, resection of the primary tumour, change in pancreastatin concentration post-treatment and tumour site all made a significant impact on outcome. Resection of the primary tumour is frequently associated with less advanced, therefore operable disease.
During- to pre-treatment pancreastatin ratio χ 2 was 24.364, P < 0.001 and tumour site χ 2 was 10.36, P = 0.006. Kaplan-Meier plots showing survival differences by change in pancreastatin concentration during treatment and primary tumour are shown in Figures 2 and 3, respectively.
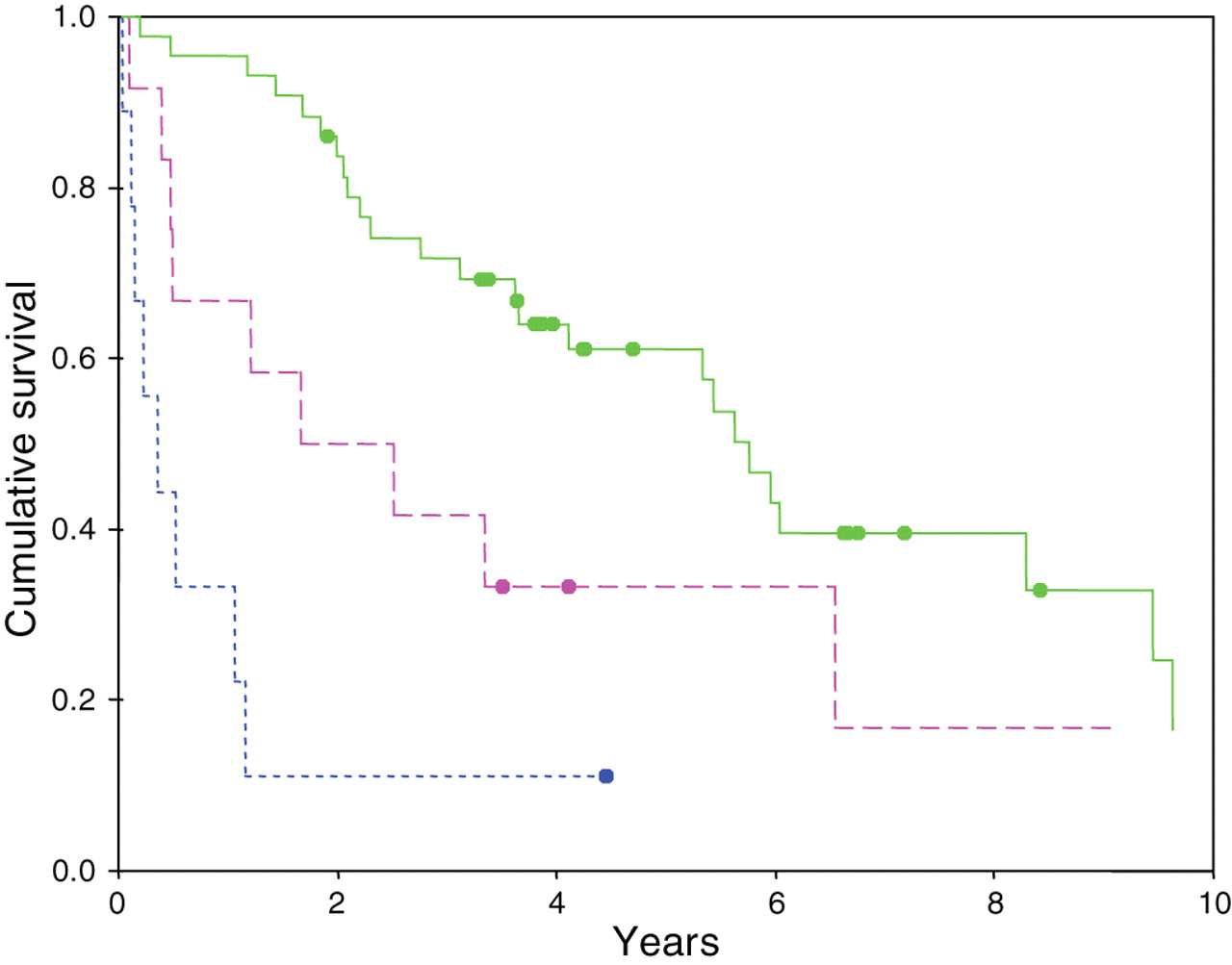
Kaplan-Meier plot of cumulative survival against the change in pancreastatin (during-treatment:pre-treatment pancreastatin ratio). Solid line denotes reduced or no change; dashed line denotes 1.5-fold to five-fold increase; dotted line denotes >five-fold increase: (blot denotes censored). χ 2 = 24.364 (P < 0.001)
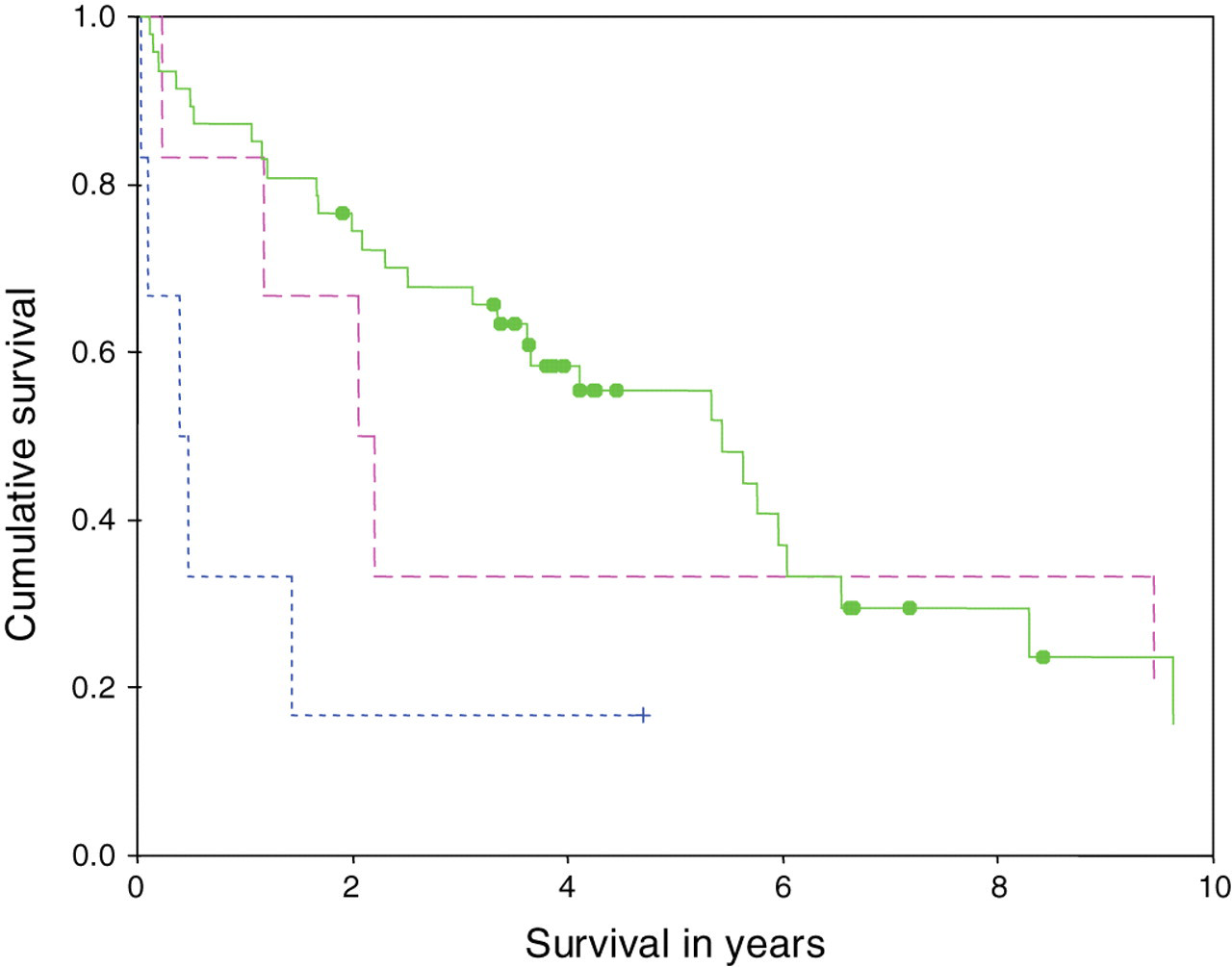
Kaplan-Meier plot of cumulative survival against primary tumour site. Solid line denotes midgut carcinoid tumours; dashed line denotes broncho-pulmonary tumours and dotted line denotes pancreatic tumours (blot = censored). χ 2 = 10.36 (P = 0.006)
Multivariate analysis
Primary tumour site, resection of the primary tumour and change in pancreastatin concentration following treatment were selected for multivariate analysis and were entered into a Cox proportional hazards model. In addition, age and pre-treatment pancreastatin concentration, although not significant in univariate analysis in this data-set, have been found to be indicators of poor prognosis in other studies and were therefore also included in the model.
There was a highly significant difference in survival outcome between the three tumour locations (P = 0.001) (Table 2). Tumours of the ileum and colon had the most favourable outcome followed by broncho-pulmonary and then by pancreatic tumours. The effect of pre-treatment pancreastatin concentration on survival was also significant (P = 0.04) with higher concentrations being associated with a poorer outcome. Change in pancreastatin following treatment was a highly significant prognostic indicator in the group (P < 0.001). An up to five-fold rise in pancreastatin during treatment indicated a significantly poorer outcome (hazard ratio [HR] 3.5) and a rise of greater than five times indicated an even poorer prognosis (HR of 17.3). The negative survival effects of increasing age or lack of surgical resection did not reach statistical significance.
Cox model results in 59 patients with neuroendocrine tumours
MGC = mid-gut carcinoid
In a further analysis, the sub-grouping of pancreastatin concentrations was removed and pre-treatment concentrations and during-treatment to pre-treatment ratios were included in the Cox regression model after both had been logarithmically transformed to render them less heavily skewed. Although pre-treatment concentrations and during-treatment to pre-treatment ratios were correlated, each made a highly significant contribution to the model independent of the other (P < 0.001). A 10-fold increase in pre-treatment pancreastatin concentration was associated with a HR of 3.6 (95% confidence interval [CI] 1.8, 7.3) while a 10-fold increase in during-treatment to pre-treatment ratio was associated with a HR of 5.5 (95% CI 3.0, 10.2).
Analysis of patients with tumours of the ileum and colon alone showed similar results for pre-somatostatin analogue pancreastatin, but the overall comparison of the three groups was no longer significant. For during- to pre-ratio ‘significant’ and ‘very significant’ rises were associated with lower HRs than before and only the latter was significant. When analysed as continuous variables on log scale, both pre-therapy pancreastatin and during- to pre-pancreastatin ratios remained significant (HRs 3.5 and 4.3, respectively, compared with 3.6 and 5.5 in the full data set). Age and surgical resection were not associated with a significant effect on survival in this subgroup.
Discussion
In this study, an elevated pre-treatment plasma pancreastatin concentration was found to be a significant indicator of poor outcome. When pancreastatin is >500 pmol/L at presentation, this is an independent indicator of poor outcome. This finding is in agreement with data from other groups both for pancreastatin and whole molecule CgA. 7,8,13 As pancreastatin is known to correlate with the number of liver metastases, it may be used as an indicator of the progression of metastatic disease in patients with NETs.
Although reports have indicated that the majority of patients with carcinoid syndrome and pancreatic NETs show a clinical response to somatostatin analogues, 14 no study to date has demonstrated a negative response to treatment. This is the first study to show a paradoxical rise in a biochemical tumour marker in response to treatment with somatostatin analogues in a group of patients with highly differentiated NETs. In the present study, 20 (40%) of the 59 patients showed a significant rise in pancreastatin within one year (1–42 weeks) of commencing somatostatin analogue treatment. This rise in circulating pancreastatin during therapy was associated with a worse survival when compared with those patients who showed positive or neutral biochemical response. This finding was substantiated by a graded response that was illustrated by the sub-groups imposed on the data, which showed that those patients with higher during- to pre-pancreastatin ratio showed poorer survival. The reasons for these findings are unclear. Some patients who showed a negative response to treatment with respect to pancreastatin had advanced disease, but likewise some who responded well also had advanced disease. Also some patients in all groups had a small amount of disease during the commencement of treatment and their response to treatment was not predictable from the tumour bulk according to radiology findings or pre-treatment pancreastatin concentrations. Nine (15%) of 59 patients who showed a fall or steady pancreastatin during treatment had pre-treatment concentrations within the reference range (survival 5.7 years in this group), three (27%) of 11 who showed a significant rise had pre-treatment concentrations within the reference range (survival 1.7 years) and four of nine patients who showed a highly significant rise had pre-treatment concentrations within the reference range (survival 0.3 years). The association between a rapidly rising circulating pancreastatin in response to somatostatin analogue treatment and poor outcome was more highly significant than the association between elevated pre-treatment pancreastatin and poor outcome. This may suggest that patients with a rapidly rising pancreastatin were in, or entered a tumour phase with rapid tumour progression. Whether this was spontaneous or in response to treatment is not clear.
It has been reported that an elevated proliferative index assessed by Ki67 indicated poor prognosis in NETs. 15 Patients with high Ki67 are expected to show a poor response to somatostatin analogues and guidelines recommend the use of other treatment modalities. 16 However, a single report of high-dose treatment with somatostatin analogues in patients with advanced midgut carcinoid tumours has shown tumour stabilization in 75% of patients with a reduction of Ki67 in 25%. 17 At the time of referral, patients frequently consent for liver biopsy. For these and for those who undergo surgery, Ki67 was assessed. However, it is uncommon if not unethical for patients to undergo repeated biopsy; therefore, Ki67 is not readily accessible for assessment of tumour change and most units rely on radiology along with tumour markers and peptide biochemistry to assess tumour progression. Ki67 had been measured in only a small number of the subjects in this study (18 of 59) within the time limits, and was raised (>3%) in only one pancreatic tumour.
Another potential mechanism for poor response to somatostatin analogue therapy is that somatostatin analogues may be implicated in the regulation of oncogenes or tumour-suppressor genes. 17 The tumour suppressor gene, Zac1, is essential for the anti-proliferative action of somatostatin. It has been shown that knocking-out Zac1 abolishes the inhibitory action of somatostatin analogues.
NETs express a high density of somatostatin receptors for which somatostatin analogues have high affinity, particularly receptor SST2. Altered regulation of receptors or the expression of different receptor subtypes in this sub-group of patients may contribute to the unexpected negative response to somatostatin analogues in some patients. All patients however had positive somatostatin receptor scintigraphy, which relies on the same receptor sub-types.
In summary, this study supports the finding that pancreastatin is a useful prognostic indicator of neuroendocrine disease. Further, in more than one-third of subjects a negative biochemical response to somatostatin analogues therapy was observed and was associated with poor survival outcome. This is the first study to identify such a correlation and should be followed up with a prospective study in order to substantiate these findings. Given that the described biochemical response can be identified at an early stage of treatment, these findings have significant therapeutic consequences and may be used to identify patients who are not responding to somatostatin analogue therapy even when symptoms are relieved. In the presence of a rapidly rising pancreastatin other treatment modalities should be sought with urgency.
Footnotes
ACKNOWLEDGEMENTS
This project was supported by a Wellcome Foundation Elective Prize and a studentship with The School of Medicine, Queen's University, Belfast.