
Abstract
Abstract
Central pontine myelinolysis (CPM) is well recognized to occur in a variety of clinical settings, but particularly following rapid correction of severe hyponatraemia. The development of CPM as a result of rapid shifts in plasma osmolality during the treatment of the hyperglycaemic hyperosmolar state (HHS) has hitherto been described in only one case. We report a second case in which this complication occurred in association with treatment of the HHS.
The patient was a 49-year-old woman who presented to another hospital with drowsiness and a plasma glucose of 106 mmol/L. Her admission plasma sodium was 135 mmol/L. She received treatment with intravenous insulin and 0.9% normal saline, and there was a rapid drop in plasma glucose by 60 mmol/L within 6 h, which was associated with a rebound rise of plasma sodium to 159 mmol/L. Her plasma glucose and sodium were later stabilized. When the patient was transferred to our hospital a few days later, she was noted to have flaccid quadraparesis and pseudobulbar palsy. A magnetic resonance imaging scan revealed a pontine lesion consistent with CPM. She made a gradual recovery over several months with intensive rehabilitation and eventually returned to near normal functional capacity. This is the second case report in the literature of CPM complicating the management of HHS and highlights the importance of the judicious and measured correction of hyperglycaemia and the appropriate management of fluid replacement and electrolyte balance when treating this condition.
Introduction
Central pontine myelinolysis (CPM) is a rare demyelinating condition which has been reported to occur in a variety of clinical settings, but most commonly in association with a rapid rise in plasma osmolality during correction of chronic hyponatraemia. 1 The clinical consequences can vary from mild motor weakness that resolves completely over time to the devastating locked-in syndrome. 2 Treatment of the hyperosmolar hyperglycaemic state (HSS) can cause major shifts in plasma osmolality with a rapid rise in plasma sodium as plasma glucose falls with hydration and insulin therapy. However, CPM occurring in the context of the treatment of HSS has hitherto been reported in only one case with fatal outcome. 3 In this paper, we report a case of a patient who developed CPM as a result of the treatment of HHS, but who later made a full functional recovery.
Case history
A 49-year-old woman with no previous history of diabetes mellitus presented to another hospital with collapse. She had a one-day history of abdominal pain and a two-month history of polydipsia and anorexia. She had a background of autoimmune hypothyroidism controlled with levo-thyroxine and schizophrenia for which she received regular depot injections of fluphenzine and oral sodium valproate. She had a 75-pack-a-year history of smoking, but her alcohol intake was moderate.
On examination, she was clinically dehydrated and drowsy with a Glasgow Coma Scale score of 9/15. She was hypotensive at 80/40 mmHg and her abdomen was distended with absent bowel sounds but without tenderness or guarding. She had no focal neurological deficits. The remainder of her clinical examination was unremarkable.
Blood glucose on admission was 106 mmol/L, but her urine revealed only a trace of ketones. Arterial blood gas showed a metabolic acidosis with a pH of 7.19, bicarbonate of 3 mmol/L, and a partial pressure of oxygen (PO2)of 10 kPa. Her partial pressure of carbon dioxide (PCO2) was 4.3 kPa. She had a serum urea of 23.3 mmol/L (reference range 2.5–8 mmol/L) and creatinine of 135 umol/L (reference range 60–120), but a normal plasma sodium of 135 mmol/L (reference range 135–145). Chest radiography showed a right lower zone infiltrate consistent with pneumonia. Given her presentation with hypotension, the most likely cause of her metabolic acidosis was septic shock.
She was treated for HHS with intravenous fluids and insulin. She received 4 L of 0.9% saline over a four-hour period during which there was a marked drop of her serum glucose by 60 mmol/L with a corresponding rise of her serum sodium from 135 to 161 mmol/L over 6 h (Figure 1). Thereafter, her fluids were changed to half normal saline. There was a further drop in her glucose to a nadir of 2.4 mmol/L over the following few hours with a corresponding rise in her plasma sodium to 166 mmol/L (Figure 1). Her potassium on admission was 2.6 mmol/L (reference range 3.5–5) and normalized with potassium intravenously. Her magnesium that was normal on admission but dropped to 0.66 mmol/L on day 2 (reference range 0.85–1.15). Her phosphate on admission was also 0.43 mmol/L (reference range 0.74–1.52). She received intravenous phosphate and magnesium supplementation. Over the following days, she became increasingly septic. She developed multiorgan failure and required intubation, assisted ventilation and renal dialysis for which she was transferred to our hospital. She was treated with broad-spectrum antibiotics, the sepsis slowly resolved and laboratory abnormalities returned to normal.
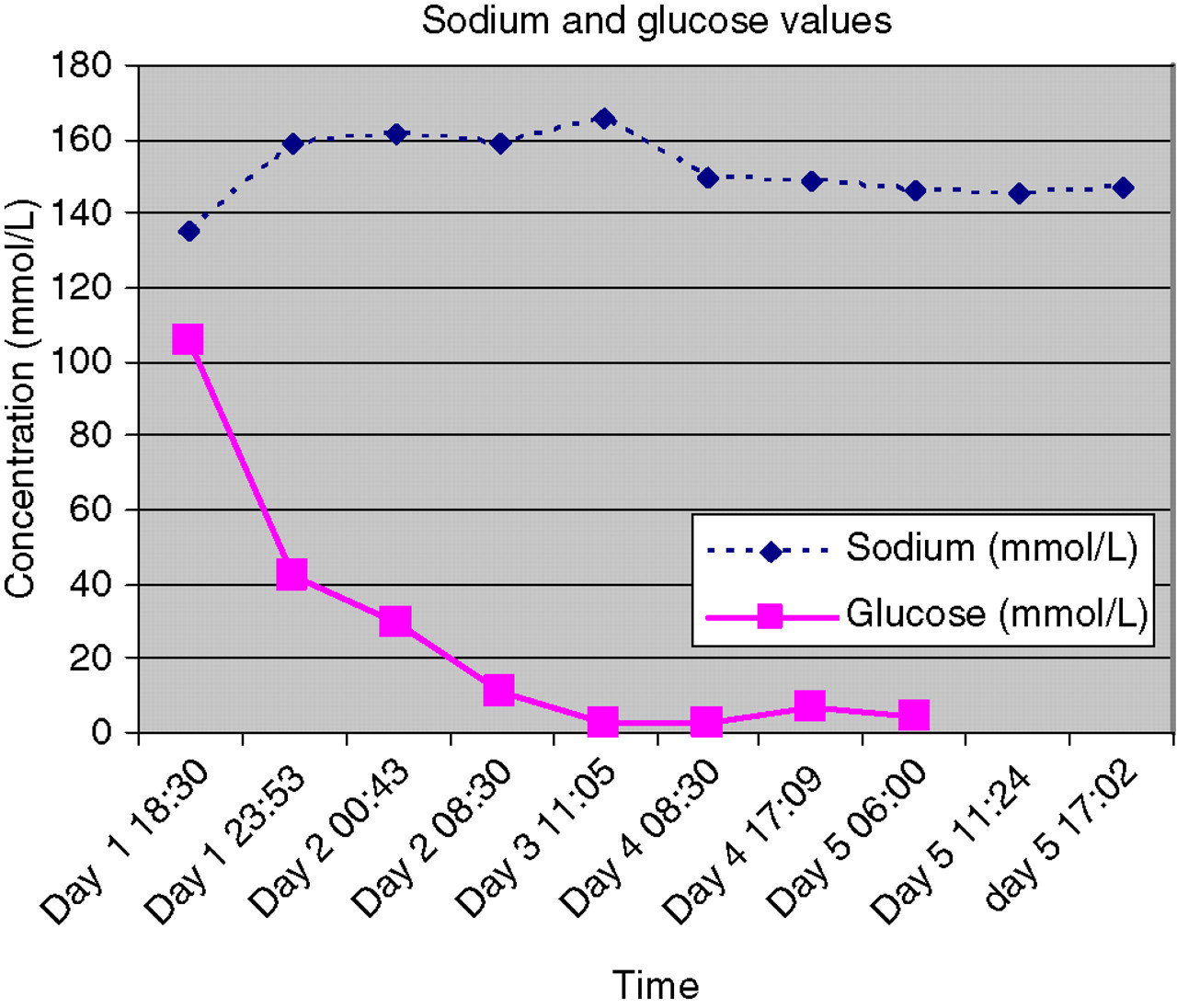
Changes in plasma glucose* and sodium concentrations during the early period after admission. *To covert plasma glucose concentration from mmol/L to mg/dL, multiply by 18.3
Sedation was stopped on day 9. Over the following few days her Glasgow Coma Scale score improved to 11/15. However, neurological examination revealed a flaccid quadraparesis with reduced tone, power and reflexes in all four limbs and a psuedobulbar palsy with dysarthria and impaired swallow. She could not tolerate tracheal extubation and had a tracheostomy tube inserted. A percutaneous endosocopic gastroscopy (PEG) tube was inserted for feeding. CPM, which was suspected, was confirmed by a magnetic resonance imaging (MRI) scan of the brain, which showed a high signal abnormality in the pontine area consistent with pontine demyelination (Figure 2).
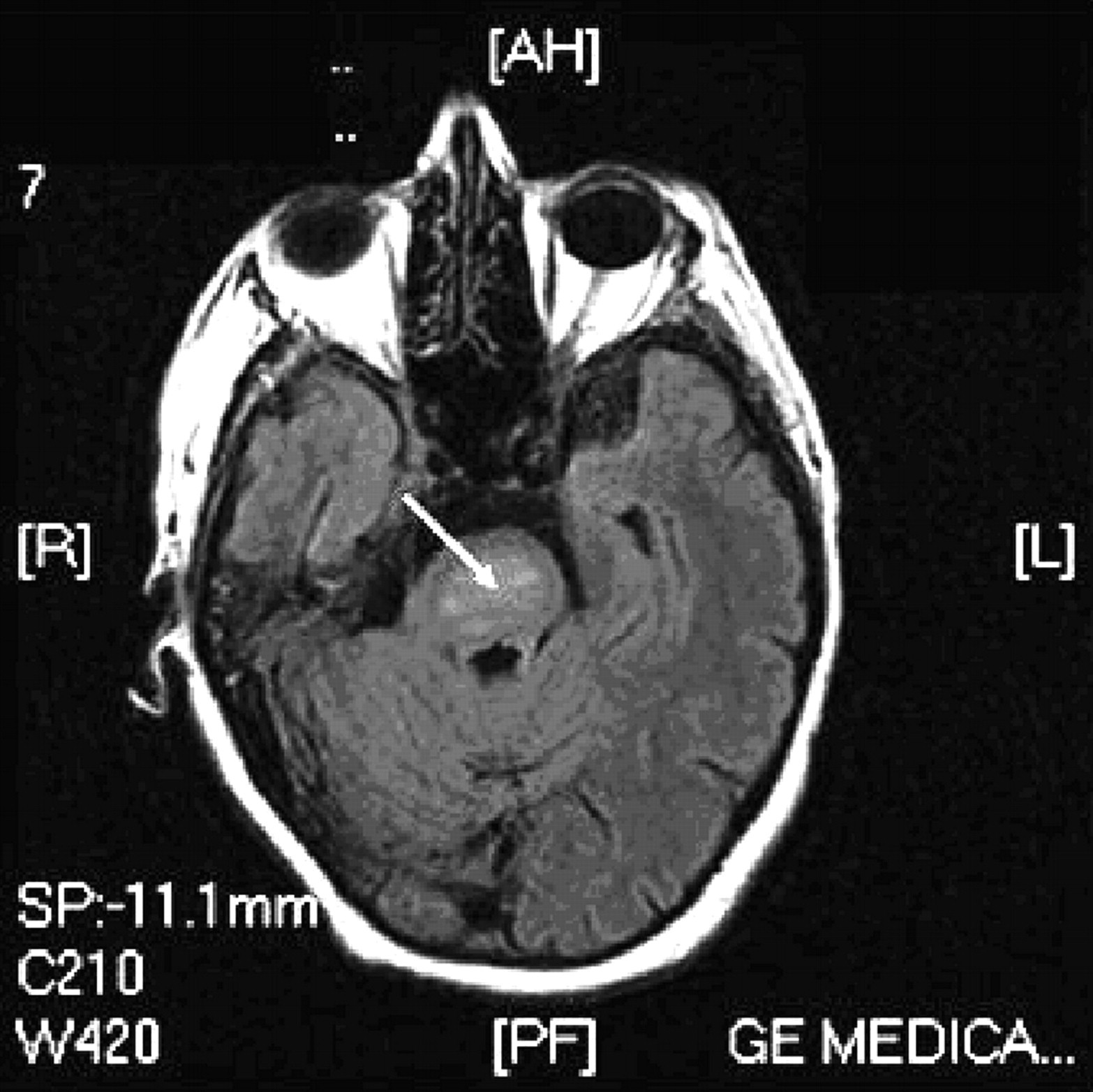
Brain magnetic resonance imaging (MRI) with flair, demonstrating high signal in the pontine area (arrow) consistent with central pontine myelinosis (CPM)
The patient underwent intensive rehabilitation and she slowly regained mobility over the course of several weeks. Her swallow, speech and respiratory function gradually recovered and the tracheostomy and PEG tubes were later removed. She was discharged from hospital 90 days following admission. At her follow-up assessment six months later, her diabetic control was excellent with metformin and her glycosylated haemoglobin was 5.7% (target <6.5%). She had almost complete functional recovery and was independent in all activities of daily living.
Discussion
We have described a case of a patient in whom the treatment of severe HHS was complicated with clinical and radiological evidence of CPM. The most likely cause for CPM in this case was the significant shift in serum osmolality as her hyperglycaemia was corrected, which was associated with a rapid rise in plasma sodium from a baseline of 135 mmol/L to a peak of 164 mmol/L. We can also speculate that malnutrition as well as deficits in total body potassium and sodium in addition to other osmolytes such as phosphate and magnesium, which occur in the setting of HHS may have contributed to the development of CPM in our patient.
CPM was first described by Adams et al. 4 in 1958. In their seminal paper, they reported four cases of patients with alcoholism and malnutrition who developed varying degrees of encephalopathy, quadriplegia and pseudobulbar palsy. At necropsy, this cohort of patients had striking pontine changes. It was not until 1976 that it was postulated by Tomlinson et al. 5 that CPM may be caused by rapid correction of chronic hyponatraemia. This theory has been verified in animal studies. Since then, it has become clear that CPM can occur in a variety of clinical settings, the most common being the rapid correction of severe hyponatraemia, 1 but it has also been described in association with other electrolyte abnormalities such as hypernatraemia and hypophosphataemia, 6,7 as well as in the setting of liver transplantation and lithium and carbamate poisoning. 8–10 This is the second reported case of CPM complicating the treatment of HHS, the first report was of a woman who presented with hyperglycaemia and hypernatraemia. Her initial plasma sodium was 169 mmol/L and increased to 188 mmol/L during correction of the hyperglycaemia and then gradually fell. She remained obtunded and died three weeks after presentation. Her postmortem showed extensive demyelination in the pontine area. 3
CPM is more common in women and can also affect extra-pontine areas (EPM) of the brain. 2 The exact aetiology of CPM is unclear. In the setting of acute hyponatraemia, there is an osmolar shift leading to cellular swelling and cerebral oedema. If hyponatraemia occurs over a prolonged period (>48 h), the cells adapt in order to maintain normal size. The first adaptive mechanism is the extrusion of sodium and potassium from the cell, which occurs over a period of hours. This is followed by organic osmolytes, such as myoinisotol, taurine and glutamate, which occurs over a period of days. 11 If the rate of correction of plasma sodium is greater than the rate at which organic osmoles can be re-synthesized or transferred back into cells, cell shrinkage occurs, ultimately leading to osmotic demyelination. In HHS, the hyperglycaemia leads to a reduction in plasma sodium concentration because of the dilutional effect caused by the shift of water from the intracellular to the extracellular compartments (translocational hyponatraemia). With treatment as the plasma glucose falls water shifts back into the intracellular space and the plasma sodium rises. Therefore in HHS, the corrected rather than the laboratory-measured plasma sodium should be calculated (Box 1). 12 If the corrected serum sodium concentration is high (in our patient corrected plasma sodium on admission was 178 mmol/L), then hypotonic fluids should be administered (0.45% saline) and plasma sodium should be carefully monitored with the rate adjusted. In addition, reduction in serum glucose should be gradual and balanced carefully against the rebound rise in plasma sodium. When the serum glucose concentration is <16.7 mmol/L, the fluid may be changed to 5% dextrose solution. 13 Adequate potassium supplementations and measures to prevent other complications such as venous thromboembolism should be instituted and these have been reviewed by Trence and Hirsch (2001) 13 and Stoner (2005). 14
Calculation of corrected plasma sodium in hyperglycaemic patients
Corrected serum Na = measured serum Na+ [ΔSG ÷ 42]
The ΔSG is the increment in serum glucose above normal if serum glucose concentration is measured in mg/dL. The ΔSG should be divided by 2.3 if glucose is measured in mmol/L.12 ΔSG, change in serum glucose.
Pathologically, CPM is characterized by symmetrical, non-inflammatory demyelinating lesions. Lesions in the pons are usually confined to the basis pontis, but may extend to the tegmentum. In EPM, the midbrain, thalamus, basal nuclei and cerebellum can be involved. The central pontine area contains an admixture of white and grey fibres organized in a tight grid-type arrangement making it particularly vulnerable to osmotic demyelination. EPM also occurs in the areas of greatest grey and white admixture. Cellular oedema caused by changing osmotic forces may lead to compression of fibre tracts and in turn causing demyelination. 15
The clinical features of CPM are highly variable. Pseudobulbar palsy due to involvement of the cortiocobulbar tracts leads to head and neck weakness as well as to dysphagia and dysarthria. Involvement of the corticospinal tracts causes quadraparesis, which is initially flaccid, becoming spastic later. Coma and confusion result from lesions in the pontine tegmentum or thalamus and a large basis pontis lesion can cause a locked-in syndrome characterized by paralysis of the lower cranial nerves and limb musculature. MRI is the investigation of choice and has been shown to be a sensitive tool in detecting CPM. 16 However, neuroimaging will not always demonstrate a lesion which highlights the importance of thorough clinical examination. Appearance of CPM on imaging may be delayed. If clinical suspicion is strong, then repeat imaging 10–14 days later is warranted.
CPM was previously considered to be a fatal condition diagnosed postmortem. However, the greater availability of neuroimaging has resulted in an increased rate of diagnosis and less severe or even asymptomatic cases are now seen. In a recent series of 34 patients with CPM, two patients died, 10 survived but were left dependant, 11 had some neurological deficit but were otherwise independent and 11 patients made complete recovery. 16 Patients who survive CPM are likely to require extensive rehabilitation and input from the multidisciplinary team.
This case illustrates to clinicians the complexity of managing patients with HHS and the importance of cautious correction of hyperglycaemia and the appropriate management of fluid replacement and electrolyte imbalance. Inappropriate treatment with too rapid a shift in plasma osmolality can unfortunately lead to the potentially devastating complication of CPM.