
Abstract
Abstract
Cardiac troponin T (cTnT) and I (cTnI) are highly specific and sensitive biomarkers of myocardial cell damage and are now accepted as the ‘gold standard’ diagnostic test for acute coronary syndrome and supersede the classical muscle enzyme biomarkers. While the understanding of the development and structure of the troponins has advanced, detailed biochemistry of the troponin molecules is complex and poorly understood. Many post-translational molecular forms of troponin are known to exist. The diversity of these circulating forms may have a clinical impact and the notion of a disease-specific troponin protein signature has been suggested. However, the effects of these multiple forms on commercial assay performance and their impact clinically are currently unknown and should be the focus of future research and assay design.
Introduction
Since their emergence in the late 1980s, cardiac troponin T (cTnT) and I (cTnI) have demonstrated a high specificity and sensitivity for myocardial cell damage and play a central role in aiding the diagnosis of acute coronary syndrome (ACS). The superior clinical value of troponin measurement over classical markers such as creatine kinase (CK) and its MB (CK-MB) isoenzyme have lead to the recommendation that they are adopted as the ‘gold standard’ cardiac biomarker following the universal definition of acute myocardial infarction (AMI). 1 Cardiac troponin measurement also offers considerable benefit for risk stratification and interventional decision-making in ACS patients. 2 A number of post-translational modifications occur to both troponin molecules that may be disease-specific. 3 It is currently unknown if these modified products have any clinical relevance and their effect on assay performance is not fully understood. This has implications for current commercial assay performance and will greatly impact on the design of future assays.
Troponin structure
The tadpole-shaped troponin complex is located on the thin filament of the contractile apparatus (Figure 1) in all types of striated (skeletal and cardiac) muscle, but is not found in smooth muscle. It is formed from three subunits: troponin T, troponin I and troponin C. Troponin T (TnT: 37 kDa) binds the complex to the tropomyosin strand of the thin filament providing structural anchorage. TnT also modulates the response to intracellular Ca2+ by enhancing the interaction between myosin and actin. 4,5 Troponin C (TnC, 18 kDa) binds calcium ions released from the sacroplasmic reticulum. Troponin I (TnI, 22.5 kDa) inhibits the actin–myosin Mg2+ dependent enzymatic hydrolysis of adenosine triphosphate (actinomyosin ATPase activity) that powers muscle contraction.
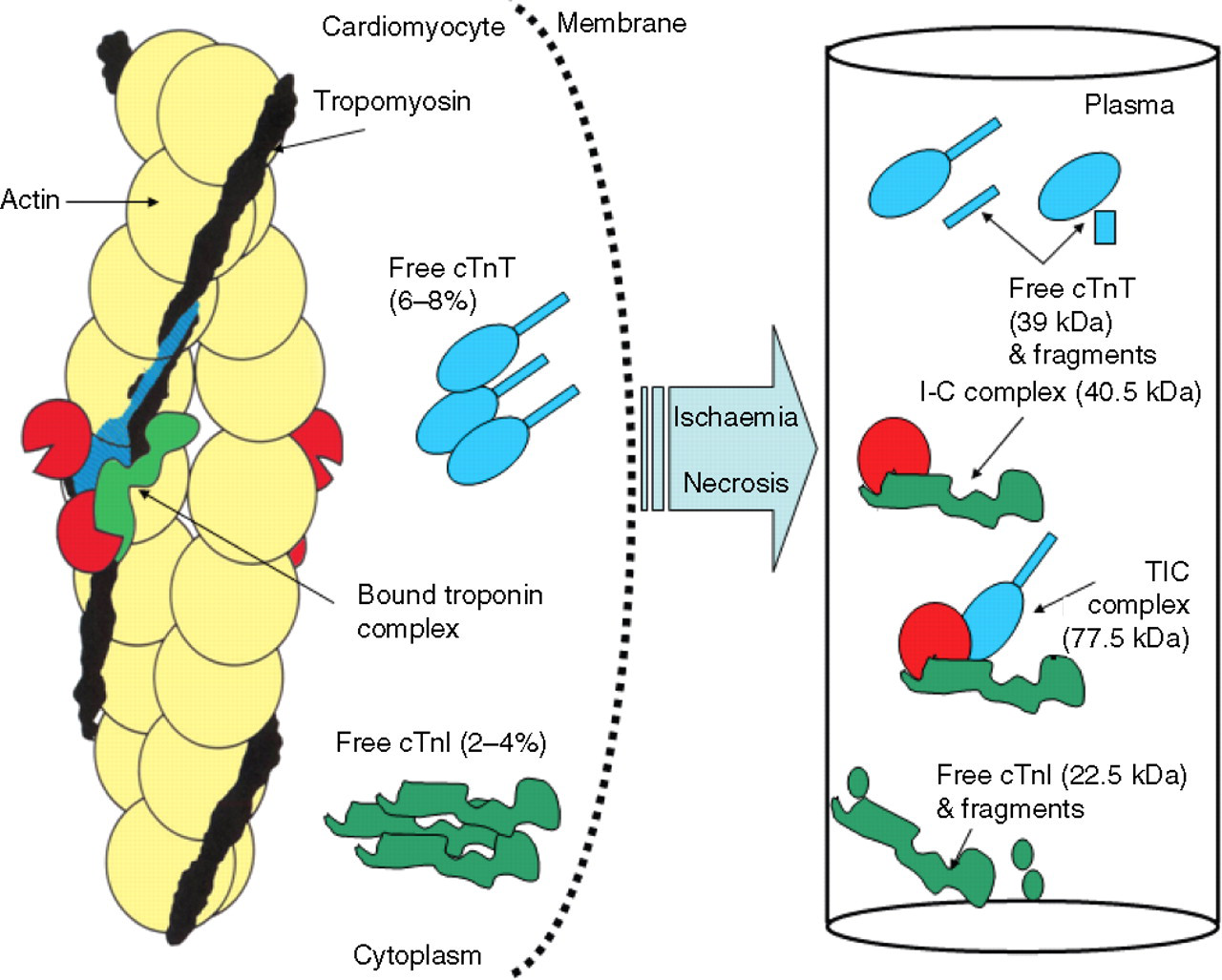
Structure of the cardiac troponin complex and troponin forms released following ischaemia or necrosis. The troponin complex is composed of three proteins: troponin T, troponin I, troponin C – TnT, TnI and TnC – and is located on the thin filament of the cardiomyocyte. Most intracellular troponin is bound to the myofibril with only 6–8% of cTnT and 2–4% of cTnI existing in the cytoplasm. Following ischaemia and necrosis, troponin from the cytoplasm and enzymatically digested bound complexes are released into the circulation. Circulating TnI exists mainly as cTnI-TnC and some cTnT-cTnI-TnC complex with little free or fragmented cTnI. The phosphorylated forms are not depicted here as it is currently not known if they exist in complex or free form
Only recently has the structure of the core domain of human cardiac troponin in the calcium saturated form been described. 6 The structure of the ternary TnT–TnI–TnC (TIC) complex implies that Ca2+ binding to TnC initiates the removal of the carboxyl-terminal portion of TnI from actin, altering the mobility and flexibility of troponin and tropomyosin on the actin filament. An in-depth review of troponin biochemistry is beyond the scope of this review and the reader is directed to the work of Dean 7 and Collinson et al. 8
Troponin ontogeny
Cardiac and skeletal muscles develop from different embryonic cell precursors, 9 but share a common developmental pathway. Many genes from both cell types are cross-expressed during embryonic development. Different forms of TnT and TnI are found in cardiac, fast twitch and slow twitch skeletal muscles. Each muscle-specific troponin type is encoded by separate genes. TnC is common to all muscle types. During the development of fetal muscle, all three TnT proteins are expressed simultaneously. A diversity of TnT isoforms could potentially exist as a result of alternative splicing of mRNA from a single gene transcript. In reality, very few spliced variants are expressed at the protein concentration. Fetal cTnT isoforms (TnTf) are transiently expressed. 10 These are down-regulated in adult cardiac tissue. 11 In human fetal skeletal muscle, a TnTf is expressed which is proportionally low in concentration. A fetal exon with sequence homology to adult fast skeletal TnT (sTnT) and cTnT has been described. 12 Fast or slow sTnT are not detectable in fetal cardiac tissue. 11,13–14 Studies have reported that the cTnT gene is expressed at low concentrations in both cardiac and skeletal muscle and expression continues until mid fetal development. At this stage, the gene is up-regulated in cardiac myocytes and suppressed in skeletal myocytes 15 initiating terminal differentiation for both cardiac and skeletal myocytes. Conversely, cTnI is not expressed during embryonic or fetal development and is only detectable in adult cardiac tissue. 16 Slow sTnI is the dominant form expressed during fetal cardiac tissue development. This is then switched to expression of cTnI during the first nine months of life. 17 This means that cTnI may be of questionable value in neonatal cardiology; 18,19 although a number of studies report elevations of cTnI in sick neonates, which probably represents true myocardial dysfunction in the absence of atheromatous disease. 20–23 The fetal heart has a lower threshold for calcium activation of contraction than does the adult heart, partly due to the changes in cTnI isoform expression. The heart has different demands placed on it through its development and alterations to contractile function are accompanied by changes in cardiac protein expression.
Functional studies have demonstrated that the different isoforms of cTnT may have varying roles in modulating contractility. 24 The shift from fetal to adult cTnT in rabbits is correlated with changes in calcium sensitivity. 25,26 In bovine myocardium, the two adult cTnT isoforms differ by five residues in the N-terminus and confer different calcium sensitivities. 27 In normal human myocardium, Anderson et al. 11 describe two adult cTnT isoforms (cTnT1 and cTnT2) expressed in different amounts, of which cTnT1 is predominant. 11 The abundance of the cTnT2 isoform is increased in myocardial tissue of patients with end-stage heart failure. Further analysis using cloned and sequenced full length cTnT1 and cTnT2 cDNA demonstrated that the cDNAs differ by inclusion of a 15- and 30-N-terminal exon in the five-prime half of the coding region that corresponds to residues 18–22 and 22–23, respectively. 13 These regions are outside of the epitope-binding sites of the Roche antibodies (see troponin Mutations section below) and so distinguishing between each cTnT form is not possible in the current immunoassay.
Cardiac troponin gene mutations
More than 60 of the 200 identified sarcomeric proteins interact with the troponin complex. Abnormalities of any of the components of the sarcomere can result clinically as restrictive cardiomyopathy dilated cardiomyopathy (DCM) or hypertrophic cardiomyopathy (HCM). It has been estimated that 50% of patients with familial HCM (fHCM) have heterogeneous genetically determined disease. 28 Mutations in the cTnI gene occur in approximately 5% of families with fHCM. 29 There are more cTnT gene mutations, which may account for up to 15% of cases of fHCM. 30
Mutations in both cTnT 31 and cTnI 32 are associated with the development of HCM (Figure 2). There are 35 known mutations in the cTnT gene. 33,34 The majority are mis-sense mutations. One entire codon deletion has been reported, located at ΔE160 in exon 12. 30 Patients with this mutation suffer from fHCM and have a poor prognosis due to sudden death with a relatively short life expectancy of approximately 35 years. A further codon deletion (ΔK210) in exon 14 produces DCM. 35 There are 27 known mutations in the cTnI gene. Twenty-five are mis-sense mutations, along with one deletion (ΔK183) responsible for HCM and one non-sense mutation (G203fs). The effect of some of the known mutations in cTnT and cTnI on calcium sensitivity, and force generation have been demonstrated, 36–38 but their effect on current immunoassay performance is unknown.
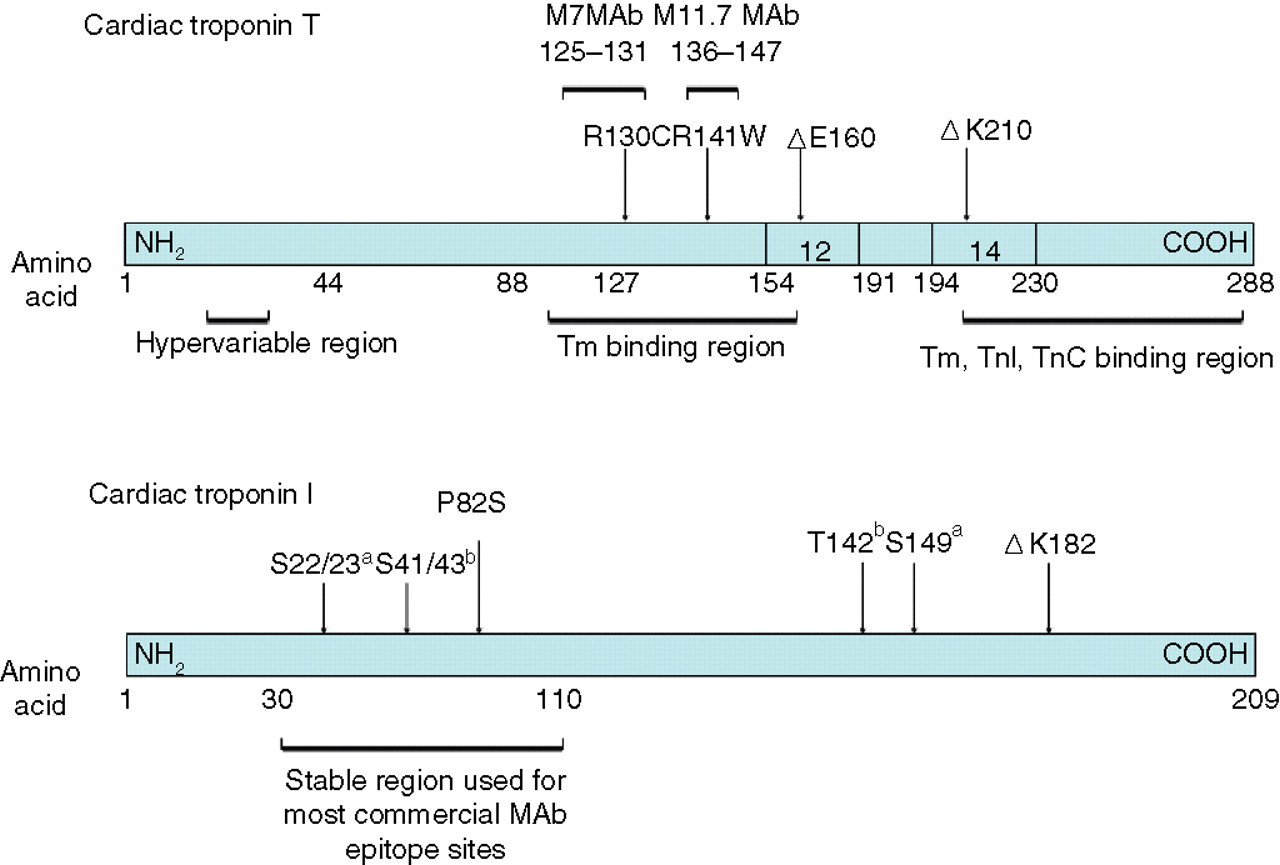
Mutations in cardiac troponins. M7 and M11.7 MAb are those employed in the Roche cTnT assay. (a) Sites phosphorylated by protein kinase A. (b) Sites phosphorylated by protein kinase C. Tm, tropomyosin, TnC, troponin C
Post-translational modifications
Phosphorylation and oxidation
Phosphorylation of troponin serves to regulate the function of the troponin-tropomyosin complex. Phosphorylation via protein kinases C (PKC) or A (PKA) causes conformational changes to the complex. This in turn affects the Ca2+ dependent contraction and ATPase activity. Swiderek et al. 39 demonstrate two cTnT phosphorylation sites that when phosphorylated, in the presence or absence of TnC and cTnI leads to a lower affinity of tropomyosin to F-actin. 40
CTnI has multiple sites for phosphorylation 41,42 via three known pathways, 3 which may result in different forms both in cardiac tissue and in serum. Phosphorylation of serine at residues 23 and 24 alters the molecular conformation. Ser-23 may be constitutively phosphorylated, but ser-24 may have more functional importance. 43 β-adrenergic stimulation via noradrenaline or isoproterenol stimulates PKA-dependent phosphorylation, increasing the rate of Ca2+ release from TnC. There are also phosphorylation sites at ser-43 and -45 and thr-144; however, the physiological effect of these phosphorylated sites is unknown. 44 TnC affects the phosphorylation of cTnI via protein kinase action. 45 cTnI contains two cystine residues at positions 80 and 97. Oxidation of the sulphydryl groups affects the interaction of cTnI with other cTn proteins. The interaction between myosin and actin is more likely when cTnI is in the de-phosphorylated form. 46
Phosphorylation of TnI reduces the affinity of calcium for TnC. PKA phosphorylation of TnI has been shown to alter the kinetics of the myosin cross-bridge at sub-maximal calcium concentrations resulting in a reduced myosin binding time. 47 This is consistent with the reduced activity of PKA in human myocardial failure. 48 Phosphorylation of TnI by PKC decreases the maximum Ca2+ stimulated actomyosin ATPase activity without altering thin filament Ca2+ sensitivity. 40
Some commercially available cTnI assays are affected by the potential oxidation of SH-groups. It is not known whether circulating cTnI occurs in the oxidised or reduced state so the effect of this phenomenon is unknown, but will in theory affect assay performance. 49
During myocardial failure, the thin filament undergoes remodelling. Changes to the thin filament composition occur and contractile function is altered in myocardial failure with changes in concentrations of expression of some proteins. Alterations in phosphorylation of contractile proteins are a result of changes in kinase and/or phosphatase activity. A large evidence base suggests that thin filament protein phosphorylation plays a major role in the development and progression of contractile dysfunction in heart failure 50 primarily mediated through PKC intracellular signalling pathways; however, the involvement of phosphatases in this process has yet to be fully investigated.
Release kinetics of troponin
The troponins are released from the myocardium into the circulation in the first few hours following a period of ischaemia or infarction. Typically, following ACS the peak in serum concentration is seen at 12–24 h. An initial cytoplasmic pool is released followed by breakdown and release of the bound myofibrillar troponin complex. 8
The dominant forms of cTnI detectable in serum (Figure 1) following ACS are non-covalent ternary complexed cTnT-I-C (TIC complex) and binary complexed cTnI-C (IC complex) although complexed cTnI-T (IT complex) and free forms are also present. 49,51–52 Circulating free cTn can be detected soon after the initial ischaemic insult and prior to the detection of complexed forms. 47 It has been proposed that cytosolic troponins are released from cardiac tissue into the extracellular compartment during ischaemia while proteolytic degradation as a result of cellular necrosis is required to liberate troponins bound to the contractile apparatus. 53 However, this is a matter of debate. 54
In addition to intact cTnT and cTnI, degradation products of both troponins are present in the serum. Eleven modified cTnI products have been reported and the number and extent of modified proteins change over the time course following the acute event. 55 Some cTnT fragments have also been detected, but these modified forms occur later. There are two possible mechanisms by which these modified proteins can occur. Cell-mediated cTn degradation can occur in human myocardial tissue subjected to ischaemic injury. Changes to cTnI, in particular proteolysis, occur within the myocardial tissue of patients who undergo elective coronary artery by-pass surgery. 56 It is not known whether changes also occur to cTnT under the same conditions. Troponin degradation products and covalent complexes similar to those isolated from ischaemic cardiac tissue have been identified in serum following myocardial infarction and include both phosphorylated and non-phosphorylated species. The modification of cardiac troponin prior to release into the circulation appears to be extensive and progressive. 55,56
Studies in rats have suggested that the selective and progressive degradation and covalent complex formation of cTnI provides valuable information about the nature and extent of ischaemia. Covalent complexes between rat cTnI1–193 and TnC1–94 and cTnT191–298 and are likely the result of isopeptide bond formation between lysine193 of cTnI and glutatmine191 of cTnT by transglutaminase activity. 57 The modification of cardiac contractile proteins in particular by oxidation, phosphorylation and proteolysis and disease-induced modifications will affect the myocardial contractility. 3
Implications for cardiac troponin assays
Owing to patent licensing, there is only one cTnT assay, which is available from Roche Diagnostics. A number of cTnI assays are available. 8 Comparison between cTnI assay platforms is difficult primarily due to a lack of a common standard, variability in antibodies used in the commercial assays and choice of epitopes. Currently, this is being addressed by the AACC Subcommittee on Standardization of cardiac TnI. 58,59 This group, together with the US National Institute for Standards and Technology, have developed a TIC complex, designated standard reference material (SRM) 2921, which has been certified as primary standard. 60 Preliminary studies have demonstrated that the use of SRM 2921 has reduced the 20-fold differences in absolute cTnI concentrations to between 2- and 5-fold differences.58,59
While there has been an improvement in harmonization of cTnI assays using the SRM2921 reference material, differences still exist. 61 These could be due to in part to variation in immunoassay detection methodology as well as the various cTnI forms which may alter epitope-binding sites when cTnI is oxidized, reduced, phosphorylated in either the free, binary and ternary complexes. 58,59,61 In addition, the different antibodies used in the commercial assays may have different sensitivities to these modified forms of cTnI further contributing to the variations in concentrations obtained using different immunoassay systems.
Cardiac troponins and renal failure
The possibility that cardiac isoforms of cTnT are expressed in diseased skeletal muscle was first suggested following a report of elevated serum cTnT without cTnI elevation in a patient with acute renal failure and rhabdomyolysis. 62 The initial report was compromised due to poor assay performance as the first generation cTnT assay used a non-specific detection (1B10) antibody 63 which had 12% cross-reactivity with sTnT.
Subsequent studies have clearly demonstrated both cTnT and cTnI are elevated in renal failure, and that such elevations carry prognostic significance. 64 Furthermore a meta-analysis confirms that elevation of serum cTnT and cTnI in renal failure offers true prognostic cardiac risk with a relative risk for all-cause mortality of 2.64 (95% CI 2.17–3.20) for cTnT (>0.1 μg/L) and a relative risk of 1.74 (95% CI 1.27–2.38) for cTnI. 65
It has been reported by Diris et al. 66 that cTnT in renal failure is due to retention of circulating fragments with molecular masses in the region of 8–25 kDa and no evidence of intact cTnT. The authors attribute this to the absence of renal function causing retention of fragments normally excreted via urine. This has been refuted 67 mainly due to the lack of information regarding specificity of the clone 4C5 anti-cTnT antibody. Furthermore, Fahie-Wilson et al. 68 have demonstrated by gel exclusion chromatography a single homogeneous peak at a position between 25 kDa and 67 kDa with no evidence of low molecular weight fragments. 68 It is currently unclear exactly what is released from the myocardium in end-stage renal disease and more importantly, by what mechanism. This requires confirmation using antibodies employed currently in commercial assays.
Troponin mutations
The importance of cTn gene mutations and their effect on the performance of analytical systems are not fully understood. Two mutations occur in the epitope-binding regions of the Roche cTnT assay. R130C is located within the M7 MAb epitope region (residues 125–131), while the point mutation R141W, which causes progression from fetal HCM to DCM, is located within the M11.7 MAb epitope region (residues 136–147). A mis-sense mutation (P82S) is also known in the stable region (residues 30–110) of cTnI. 34 In theory, these mutations might cause an analytical problem. In practice, the clinical picture is dominated by the underlying disease and the ability to detect cTnT or cTnI is of relatively little concern. Mutations within the stable region of cTnI, altering phosphorylation sites, might therefore affect immunogenicity.
Post-translational modifications
Studies have demonstrated the altered immunoreactivity of complexed, phosphorylated and oxidized troponin forms using commercially available antibodies which will have implications in assay performance. 49,69–71 The standard detection of cardiac troponin in the clinical biochemistry laboratory is by immunoassay. Phosphorylation alters the overall protein charge normally resulting in reduced antibody affinity. Due to the complexity of antibody pairs and a lack of standardization, it is currently not known what commercial assays actually detect. This is, however, an area of great interest. Different phosphorylation stages reflect alterations to the contractile function of the heart. If assays were created to detect such subtle changes in troponin, there could be scope for risk stratification and alteration in clinical management of such patients.
Effects of anticoagulant sample tubes on cardiac troponin assays
The urgency of cardiac biomarker testing warrants the use of plasma rather than serum to reduce the overall turn around time from blood draw to result. The effect of anticoagulants has been shown to affect the detection. This is important both analytically in terms of assay detection and sample turn around time, but also clinically due to the use of heparin as a pharmacological therapy in patients with ACS. The recommendations for assay specifications set by the International Federation of Clinical Chemistry (IFCC) Committee; Scientific Division's Committee on Standardization of Markers of Cardiac Damage in 2001 identified the need to understand the effects of anticoagulants on binding of cTnI. 71 cTnT interacts differently with heparin than cTnI. In a study of 32 patients undergoing coronary artery bypass grafts, Septh et al. 72 demonstrated a decrease in both cTnT and cTnI in heparin plasma samples, despite the addition of protamine and heparinase to reverse the heparin effect. 72 The decrease in cTnT is postulated to be a result of direct interaction between negatively charged glycosaminoglycan and basic resides within the cTnT protein. The decrease in cTnI is indirect, however, possibly because of changes within the sample matrix per se. 72 It has also been demonstrated that the effect of heparin varies according to the antibodies used. 73
Recognition of free cTnT appears to be affected by heparin in a concentration-dependent manner. Similarly, EDTA can cause alterations in antibody binding by alterations in calcium-mediated cTnI-TnC binding, enhancing or attenuating stearic hindrance. Degradation will directly affect detection if one epitope of a dual antibody pair is affected. Proteolysis of cTnI results in many degradation peptides each with varying stability. Early work by Katrukha et al. 74 suggest that the majority of the proteolytic activity is directed at the N and C termini of the molecule. 74 The authors identified a stable part of the molecule at resides 30–110. In routine clinical practice, proteolytic cleavage does not present as a great problem as most commercial assays now use antibodies directed against the stable part of the molecule. 8 A study of 231 paired samples by Gerhard et al. 75 demonstrated reduced concentrations of both cTnT and cTnI in lithium heparin plasma when compared with serum. 75 They also demonstrated lower recoveries in the early phase rather than the late phase of the MI event. This is of critical importance due to the heavy reliance on diagnostic and prognostic decision-making in the initial hours after onset of chest pain. More recently, in studies for both cTnT 76 and cTnI, 77 similar in vitro concentrations have been reported in serum and plasma; however, in the study by Wang et al. 77 citrate anticoagulant rather than heparin was used for cTnI determination. More importantly, in the 96 matched serum plasma samples studied by Dominici et al. 76 there were no misclassifications, if plasma was used instead of serum.
Conclusion
As cTn testing is now recommended by National Service Frameworks 78 and therapeutic guidelines, 79 it is paramount that further research is directed to understand the clinical relevance of modified troponin proteins. Simply measuring cTn concentrations for AMI is only the tip of the iceberg. The identification and understanding of diseased-induced contractile protein modifications and their clinical significance will have an ultimate impact of future assay design.