
Abstract
Abstract
Background
Since sphingosine-1-phosphate (Sph-1-P) plays an important role as an extracellular mediator through interaction with specific cell surface receptors, especially in the area of vascular biology and immunology/haematology, determination of its plasma concentration may become important from the clinical viewpoint. Thus, we attempted to develop a method of measuring the plasma Sph-1-P concentration for use in the clinical laboratory setting.
Methods
After two-step lipid extraction, Sph-1-P was coupled with o-phthaldialdehyde, and the resultant fluorescent derivative was separated by high-performance liquid chromatography. C17-Sph-1-P was used as the internal standard, instead of dihydrosphingosine-1-phosphate, which had been used previously for the same purpose but was actually detected in plasma.
Results
Our procedures for preparing the plasma samples and assay Sph-1-P were found to be satisfactory for clinical laboratory testing. The plasma Sph-1-P concentrations were significantly higher in men (413.1 ± 52.0 nmol/L; mean ± SD) than in women (352.4 ± 39.7 nmol/L). Unexpectedly, strong positive correlations were found between the plasma Sph-1-P concentration and red blood cell (RBC)-related parameters, rather than platelet-related parameters.
Conclusions
Our present study confirmed the possibility of the clinical introduction of plasma Sph-1-P measurement, and in addition, suggested that RBCs may be involved in the regulation of plasma Sph-1-P concentrations.
Introduction
Sphingolipids have been clearly implicated as modulators of membrane signal transduction systems, and firmly established as being involved in diverse cellular processes. Sphingosine-1-phosphate (Sph-1-P) is one of the bioactive sphingolipid mediators, and is unique in that it functions as both an intracellular and an extracellular messenger. Among others, this bioactive lipid regulates a variety of biological functions, including cell proliferation, survival, migration and contraction. 1–4 Although the mechanism of the intracellular actions of Sph-1-P remains to be clarified, it is now established that its extracellular actions are mediated through interaction with a family of G protein-coupled receptors named S1P1–5. 1–6
It has been recognized that Sph-1-P is abundantly stored in platelets because of the high activity of sphingosine kinase and the lack of Sph-1-P lyase in these anucleate cells. 7–9 The stored Sph-1-P is released extracellularly upon stimulation by protein kinase C activators. 7,8,10 Because of the well-known expression of S1Ps in (cardio) vascular tissues, it was considered reasonable to assume that Sph-1-P may act as an intercellular lipid mediator, playing an important role in vascular biology. Indeed, a number of related studies have been reported, and the critical role of Sph-1-P in the development of the vascular system in vivo was revealed in S1P1-deficient mice. 11,12 Homozygous S1P1 −/− mice exhibited embryonic haemorrhage followed by intrauterine death at 12.5–14.5 days of gestation. 11,12
Another important area in which Sph-1-P has attracted a great deal of attention is in the field of immunology/haematology. Sph-1-P involvement in the circulation of lymphocytes between the blood and secondary lymphoid tissues has now been established; Sph-1-P interaction with S1P1 is essential for lymphocyte egress from the secondary lymphoid tissues to the lymph. 13 To support this, a marked decrease in the number of circulating lymphocytes in the peripheral blood was observed following S1P1 downregulation by FTY720 phosphate, converted by sphingosine kinase 2 in vivo from FTY720, a new immunomodulator derived from ISP-I. This novel drug, at doses that induce a marked decrease in the peripheral blood lymphocyte count, has been shown to be an effective immunosuppressive agent in experimental models of transplantation and autoimmunity and is currently undergoing phase III clinical trials for the treatment of multiple sclerosis. 14
Since growing evidence has been accumulated to indicate the important (patho)physiological role(s) of Sph-1-P, especially in the area of vascular biology and immunology/haematology, as described above, determination of the plasma Sph-1-P concentration in vivo might become important for clinical diagnostic purposes. In this study, we have proposed a method for the measurement of plasma Sph-1-P in the clinical laboratory testing.
Materials and methods
Materials
D-erythro-Sph-1-P and D-erythro-DHSph-1-P (dihydrosphingosine-1-phosphate) were purchased from Biomol (Plymouth Meeting, PA, USA), while D-erythro-C17-Sph-1-P was procured from Avanti Polar Lipid, Inc (Alabaster, AL, USA). High-performance liquid chromatography (HPLC)-grade methanol, o-phthaldialdehyde (OPA), 2-mercaptoethanol and all organic solvents for lipid extraction were obtained from Wako Pure Chemical Industries (Osaka, Japan).
Plasma sample preparation
Blood samples were collected from the antecubital vein of healthy adult volunteers who had not received any medication during the previous seven days and had given informed consent. The study was approved by the Institutional Research Ethics Committee of the Faculty of Medicine, the University of Tokyo. The whole blood samples were collected into vacuum tubes containing 3 mg/mL of ethylenediamine tetraacetic acid dipotassium salt 2H2O (EDTA), mixed with 10% v/v of citrate-theophylline-adenosine-dipyridamole (CTAD)
15
(BD Biosciences, Tokyo, Japan) or 3 mg/mL of EDTA + 10% v/v of CTAD. The anticoagulated samples were centrifuged at 2500
Lipid extraction
Lipids, including Sph-1-P, DHSph-1-P and C17-Sph-1-P, were extracted by a two-step extraction method originally reported by us,
16,17
with some modification; this lipid extraction method serves to separate Sph-1-P from most interfering phospholipids and sphingolipids. The plasma samples (200 μL) were diluted with 250 μL of physiological saline. As an internal standard, 50 μL of 2 μmol/L C17-Sph-1-P was added to the mixture. Three millilitres of ice-cold chloroform/methanol (1:2) was added to the samples, followed by thorough mixing and sonication for 30 min. Phases were separated by adding 2 mL of chloroform and 2.1 mL of 1 mmol/L KCl, followed by the addition of 100 μL of 3 N NaOH for alkalization. After centrifugation at 1400
Sphingosine-1-phosphate assay
The HPLC system employed for the Sph-1-P assay consisted of a model LC-10A liquid chromatograph (equipped with a LC-10ADVP pump, a CTO-20A column oven, an RF-10AXL spectrophotometric detector and an SIL-10ADVP auto-injector) and an SCL-10AVP integrator (Shimadzu, Kyoto, Japan). A derivatization mixture of 10 mg OPA, 200 μL ethanol, 10 μL β-mercaptoethanol and 10 mL of a 3% boric acid solution adjusted to pH 10.5 with potassium hydroxide was prepared. Then, 30 μL of this derivatization mixture was added to the lipid fraction extracted as described above, resolved into 150 μL of methanol, and left to stand at room temperature for 30 min. One hundred microlitres of the resultant derivative was injected into the HPLC apparatus. Fluorescence was monitored at an emission wavelength of 455 nm and an excitation wavelength of 340 nm after separation on a TSKgel ODS-80TM column (250 × 4.6 mm i.d., Tosoh, Tokyo, Japan) kept at 40°C. The flow rate was adjusted to 0.8 mL/min with 80% methanol in 0.05 mmol/L K2HPO4/KH2PO4 (pH 7.5).
Quantitative analysis was based on the peak heights in calibration curves prepared with known amounts of Sph-1-P and the DHSph-1-P standard and equal amounts of the internal standard C17-Sph-1-P.
Serotonin measurement by high-performance liquid chromatography
Serotonin in plasma samples was separated by HPLC with a column-switching system, and was specifically converted into a fluorescent derivative with benzylamine for convenient detection, as described previously. 18
β-thromboglobulin assay
The antigen concentration of β-thromboglobulin (β-TG) in the plasma samples was determined by an enzyme immunoassay using an Asserachrom β-TG (Roche Diagnostics, Tokyo, Japan), in accordance with the manufacturer's instructions.
Statistics
Where indicated, the statistical significance of the differences between two groups was determined by a paired Student's t-test. The correlations were evaluated by linear regression analysis. P < 0.05 was considered to denote significance.
Results
Establishment of the sphingosine-1-phosphate assay system
In this study, precolumn OPA derivatization, HPLC separation and quantification by fluorescence detection were performed for the Sph-1-P assay. Accordingly, the stability of the OPA derivatives of Sph-1-P and the related compounds was an important issue, as pointed out in the previous report. 19 In fact, the relative fluorescence intensities of the OPA derivatives of C17-Sph-1-P, Sph-1-P and DHSph-1-P gradually diminished when the OPA derivatization reaction was performed at one time-point and the resultant solution was separated into aliquots, which were then sequentially injected into the HPLC apparatus (Figure 1a, left). The degree of fluorescence reduction was quite similar for C17-Sph-1-P and Sph-1-P, while it was less pronounced for DHSph-1-P. As expected, such fluorescence reduction was not observed when the OPA reaction time was strictly kept at a constant (Figure 1a, right), as in this study. It seems likely that the physicochemical characteristics of C17-Sph-1-P are similar to those of Sph-1-P, while those of DHSph-1-P may differ slightly.
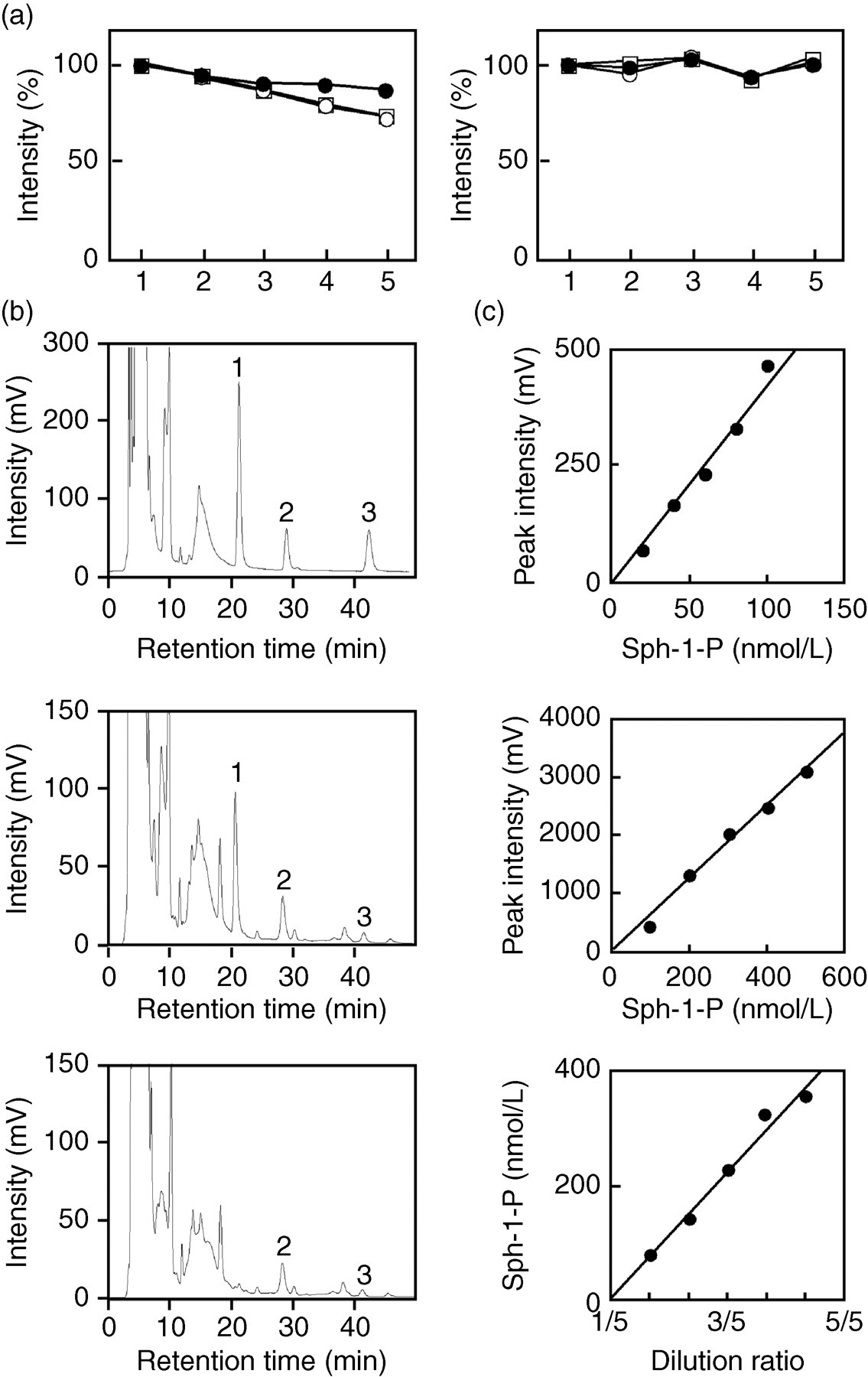
Establishment of a sphingosine-1-phosphate (Sph-1-P) assay method for use in clinical laboratory testing. (a) Stability of the o-phthaldialdehyde (OPA) derivative of C17-Sph-1-P (open circles), Sph-1-P (open squares) and dihydrosphingosine-1-phosphate (DHSph-1-P) (solid circles). (Left panel) The OPA derivatization reaction was performed once, and the resultant solution was separated into aliquots, which were then sequentially injected into the high-performance liquid chromatography apparatus. The order of injections is shown in the horizontal axis; the interval between the injections was about 1 h, and the OPA reaction times were longer for the later aliquots. (Right panel) The program was set so that the OPA reaction time was constant. (b) Chromatograms of the calibration solution (upper), plasma extract from a healthy volunteer in the presence (middle) and absence (lower) of C17-Sph-1-P. Peaks 1, 2 and 3 represent C17-Sph-1-P, Sph-1-P and DHSph-1-P, respectively. (c) Linearity of the present Sph-1-P assay. The calibration solution for 20–100 nmol/L Sph-1-P (upper), that for 100–500 nmol/L Sph-1-P (middle) and the serum sample (lower) were analysed for the linearity study
The peaks of the OPA derivatives of Sph-1-P and the related compounds could be detected in the chromatogram of the calibration standards (Figure 1b, upper). The peaks detected at the retention times of 21.2, 28.9 and 42.3 min were identified as C17-Sph-1-P, Sph-1-P and DHSph-1-P, respectively. When a plasma extract of a healthy volunteer was analysed in the presence (Figure 1b, middle) and absence (Figure 1b, lower) of C17-Sph-1-P, not only Sph-1-P but also DHSph-1-P was detected; all peaks were confirmed by analysing the samples after the addition of standards (data not shown). We detected DHSph-1-P at a concentration of 88.2 ± 28.0 nmol/L (mean ± SD, n = 30) in the plasma of the healthy subjects, which is consistent with the report on serum DHSph-1-P analysis. 20 Although DHSph-1-P has been used as the internal standard for Sph-1-P assay previously, 17,19 it seems that this compound may actually not be suitable for this purpose. Instead, C17-Sph-1-P may be an ideal internal standard, because its physicochemical characteristics closely resemble those of Sph-1-P (see above) and this compound never occurs in vivo. Hence, we employed C17-Sph-1-P as the internal standard in this study, as was the case with previous studies. 20,21
To test for the linearity of the present assay, Sph-1-P calibration solutions (0, 20, 40, 60, 80, 100, 200, 300, 400 and 500 nmol/L) and plasma samples diluted with saline were prepared. Linear relationships were confirmed between the peak heights of Sph-1-P and the concentrations in the range of 0–500 nmol/L (Figure 1c, upper and middle). Similar linear relationships were confirmed between the concentrations of Sph-1-P in the plasma samples and those in up to five serial dilutions of the samples (Figure 1c, lower). The lower limit of detection for Sph-1-P was calculated to be 0.32 nmol/L, at a signal-to-noise ratio of 10.
For the within-day and between-day assay precision tests, the Sph-1-P calibration solution (100 nmol/L) and plasma samples were used. The values of within-day assay coefficients of variation (CV) for the Sph-1-P concentrations in the calibration solution and plasma were <4.0 and 2.4% (n = 10), respectively, while the between-day assay CVs for Sph-1-P were <1.7 and 7.6%, respectively (n = 5).
Two plasma samples were used to test for recovery. Twenty microlitres of the calibration solution (1000 nmol/L) was added to 180 μL of plasma. The mean of the Sph-1-P recovery data using two plasma samples was 104%.
It was concluded that the plasma Sph-1-P concentrations could be determined reliably with this method, based on the linear relationships, sensitivity, precision and recovery data obtained.
Preparation of suitable plasma samples for the sphingosine-1-phosphate assay
Usually, EDTA is used as the anticoagulant for the preparation of plasma samples. However, extreme care should be paid to the preparation of plasma samples when platelet releasates are to be measured. For example, β-TG, a platelet-specific CXC chemokine stored in and released from the platelet α-granules, is a useful marker for the detection of platelet activation in vivo, to identify patients at risk of thrombosis and for assessing the effects of antiplatelet therapy. In the evaluation of this substance, to avoid an undesired in vitro increase in the plasma β-TG concentration, it is important to use the antiplatelet cocktail CTAD 22 as the anticoagulant, and all the plasma preparation steps should be conducted at 4°C. 22 Although EDTA blocks the coagulation cascade and platelet aggregation resulting from the αIIbβ3 integrin interaction with fibrinogen, this chelator cannot suppress intracellular signalling events leading to platelet activation, which can be effectively blocked by CTAD. 15 We examined whether these precautions would also be necessary in the preparation of plasma samples for Sph-1-P measurement, since this bioactive lipid is also abundantly stored in platelets and released extracellularly upon activation of the cells. 7
We first sought the suitable antiplatelet/anticoagulant agents for the preparation of samples for Sph-1-P assay. As expected, the serum Sph-1-P concentrations were much higher than the plasma concentrations; platelets were fully activated in the serum samples. Use of EDTA, CTAD or EDTA + CTAD greatly reduced the Sph-1-P concentration (Figure 2a). Unexpectedly, the plasma concentration of Sph-1-P was not affected by the choice of anticoagulant agent, i.e. EDTA vs. CTAD (Figure 2a); on the other hand, the concentrations of β-TG were much higher in plasma samples prepared using EDTA than in those prepared using CTAD. 22
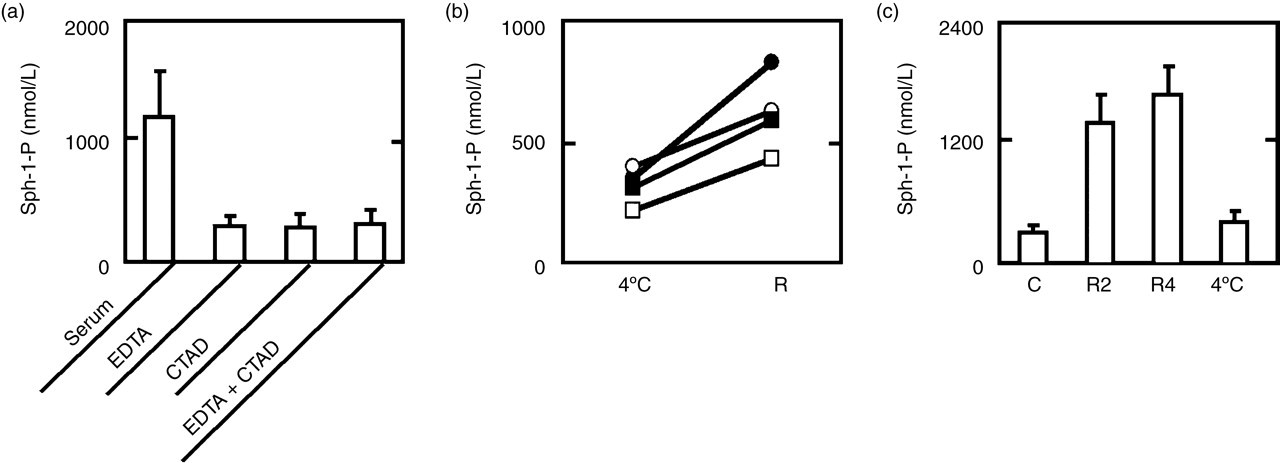
Establishment of the sample preparation conditions for the plasma sphingosine-1-phosphate (Sph-1-P) assay. (a) Effects of anticoagulant/antiplatelet agents. Whole blood samples were mixed with EDTA (E), citrate-theophylline-adenosine-dipyridamole (CTAD) (C) or EDTA + CTAD (E + C) immediately after they were collected. Then, plasma samples were prepared by centrifugation at 4°C and subjected to the Sph-1-P assay. Results are means ± SD of the data obtained from four healthy donors. (b) Effects of cooling/no cooling during centrifugation for the plasma preparation. Whole blood samples were mixed with EDTA + CTAD immediately after they were collected. Then, plasma samples were prepared by centrifugation at 4°C (4°C) or at room temperature (R), and subjected to the Sph-1-P assay. Blood samples from four healthy donors were examined. (c) Effects of incubation at 4°C or room temperature before centrifugation for the plasma preparation. Whole blood samples were mixed with EDTA + CTAD immediately after they were collected. They were then left to stand at room temperature for 2 h (R2) or 4 h (R4) or at 4°C for 4 h (4°C), and plasma samples were prepared by centrifugation at 4°C and subjected to the Sph-1-P assay. Results are means ± SD of the data obtained from four healthy donors
We next compared the concentrations of Sph-1-P in plasma samples prepared by centrifugation at 4°C and by centrifugation at room temperature; all samples were treated with EDTA + CTAD (Figure 2b). The plasma concentrations of Sph-1-P were significantly higher when the samples were centrifuged at room temperature: 319.5 ± 76.3 nmol/L in samples obtained by centrifugation at 4°C vs. 618.0 ± 163.1 nmol/L in samples obtained by centrifugation at room temperature.
We finally examined the effects of incubation at 4°C or room temperature (before centrifugation for the plasma sample preparation) on the plasma Sph-1-P concentration. Allowing the whole blood samples to stand at room temperature before the centrifugation resulted in much higher concentrations of plasma Sph-1-P, whereas keeping them at 4°C did not result in higher concentrations (Figure 2c). Accordingly, keeping the samples at 4°C through all the steps of the preparation is necessary in the preparation of suitable plasma samples for Sph-1-P assay.
Plasma sphingosine-1-phosphate measurement in healthy subjects
By employing the Sph-1-P assay and plasma preparation methods described above, we measured the plasma Sph-1-P concentrations in 30 (17 men and 13 women) healthy subjects. The mean ± SD of plasma Sph-1-P concentration in the 30 healthy subjects was 386.8 ± 55.5 nmol/L. The plasma Sph-1-P concentration was not correlated with age (Figure 3a), but it was significantly higher in men (413.1 ± 52.0 nmol/L) than in women (352.4 ± 39.7 nmol/L) (Figure 3b). Furthermore, the serum Sph-1-P concentrations were much higher but well correlated with the plasma Sph-1-P concentrations (r = 0.700, P < 0.05) (Figure 3c).
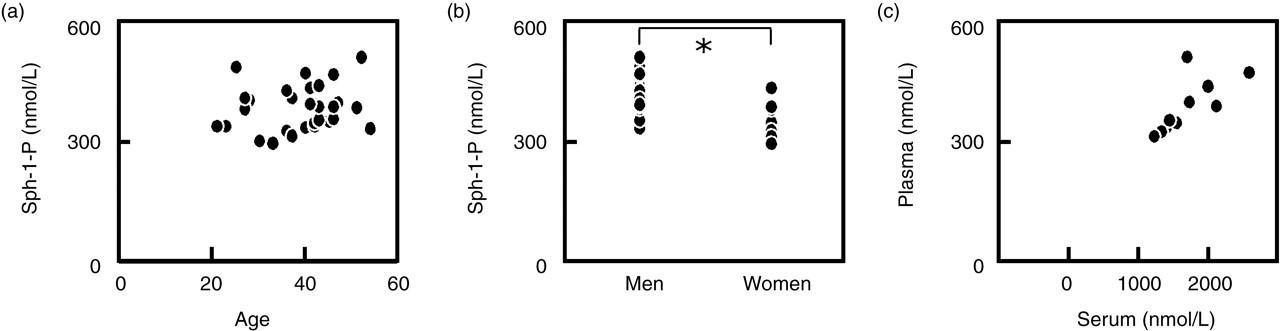
Correlations between the plasma sphingosine-1-phosphate (Sph-1-P) concentration and the age (a), sex (b) and serum Sph-1-P concentration (c) in healthy subjects
We then measured other parameters in these healthy subjects, and determined the correlations with the plasma Sph-1-P concentrations. In view of the fact that platelets abundantly store and release Sph-1-P, we had expected good correlations between the plasma concentrations of Sph-1-P and platelet-related parameters, such as the platelet count and plasma serotonin and β-TG concentrations, the latter two being well-known in vivo platelet activation markers. 23,24 No correlations were found, however, between the plasma Sph-1-P concentrations and the platelet count or plasma β-TG, with only a very weak correlation being noted with the plasma serotonin concentration (Table 1). On the other hand, the plasma concentrations of Sph-1-P were strongly correlated with red blood cell (RBC)-related parameters, i.e. the RBC count, haematocrit and serum haemoglobin (Table 1 and Figure 4).
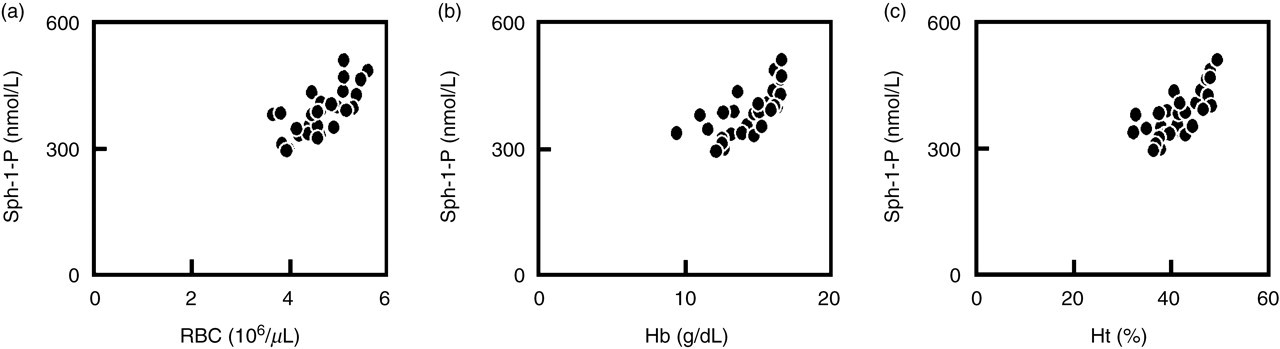
Correlations between the plasma sphingosine-1-phosphate (Sph-1-P) concentration and the peripheral red blood cell (RBC) count (a); haemoglobin (Hb) concentration (b); and haematocrit (Ht) (c)
Correlation between the plasma sphingosine-1-phosphate concentration and various parameters
The number examined was 30
It has been reported that Sph-1-P is concentrated in lipoprotein fractions in the rank order of HDL > LDL > VLDL> lipoprotein-deficient plasma, when expressed as the amount per unit amount of protein, among the plasma and serum components. Furthermore, it has been reported that the effects of HDL may be mediated, in part, by the lipoprotein-associated Sph-1-P. Accordingly, the correlations between the plasma concentrations of Sph-1-P and the lipid profile were analysed (Table 1). It was found that the plasma Sph-1-P concentrations were well correlated with the serum total and free cholesterol and phospholipid, which had been predicted to some extent. Unexpectedly, however, close correlations were found with the serum LDL-cholesterol and apolipoprotein B, but not with the serum HDL-cholesterol or apolipoprotein AI or AII.
Discussion
Based on the known interactions of Sph-1-P with blood cells and vascular cells and the established relevance of Sph-1-P in the area of vascular biology and immunology/haematology, information on the plasma concentrations of Sph-1-P and the factor(s) regulating them may give insight into the (patho)physiology of some important diseases. Our goal in the present study was to introduce a plasma Sph-1-P assay for clinical laboratory testing. This task, however, was not expected to be an easy one. One of the problems was to devise a method for reproducible measurements of the plasma Sph-1-P using conventional laboratory equipments. In addition to HPLC-based methods like our own, mass spectrometry, 25 radioactive isotope labelling techniques, 16 and competitive binding assays using cells overexpressing Sph-1-P receptors 26 have also been reported for Sph-1-P assay. It is difficult to introduce mass spectrometry into laboratory testing, and furthermore, highly quantitative analysis cannot be expected with this method. Methods requiring radioactive isotopes are also difficult to introduce for routine assay, while binding studies using intact cells may lack reproducibility and call for special facilities. HPLC-based methods are comparatively simple and highly specific, and were considered to be the best option for clinical laboratory testing of Sph-1-P. Our present method is based on a two-step lipid extraction method, originally reported by us, followed by Sph-1-P conversion to fluorescent isoindol derivatives by OPA and separation by HPLC, originally reported by Ruwisch et al. 17 To minimize variations in the results, a suitable internal standard was to be employed, and DHSph-1-P has been used previously for this purpose, since this sphingolipid possesses similar extraction properties to Sph-1-P. However, it was recently reported that DHSph-1-P also occurs in vivo, 27 and de novo biosynthesis of DHSph-1-P by sphingosine kinase 1 in mammalian cells has been reported. 28 In fact, we detected DHSph-1-P at a concentration of about 90 nmol/L in the plasma of healthy subjects. Consequently, we employed C17-Sph-1-P as the internal standard, which has not been reported to occur in vivo. Under our Sph-1-P assay conditions, the results on within-run and between-run precision, the detection limit and the linearity were satisfactory, and we considered that the present Sph-1-P assay is suitable for use in the clinical laboratory setting.
Another important problem that needed to be addressed was the preparation of suitable plasma samples for the Sph-1-P measurement. Great attention should be paid to the preparation of ‘pure plasma’ samples, i.e. plasma without platelet releasates, because platelets are easily activated during handling or centrifugation of specimens. This precaution is especially valid in the assay of substances that are abundantly stored in the platelets, including Sph-1-P. A well-known example in this context is the measurement of the platelet α-granule-derived CXC chemokines, β-TG and platelet factor 4; use of the antiplatelet cocktail CTAD, which potently suppresses platelet intracellular signalling events, 15 and preservation of the samples at 4°C were necessary to avoid in vitro platelet activation and accurately assay the plasma concentrations of these CXC chemokines. 22 Therefore, we checked the necessity of careful blood processing for precise measurement of the in vivo plasma Sph-1-P concentration. We determined that preservation and centrifugation of the platelets at 4°C were necessary as suitable preparation procedures for the plasma samples to avoid undesired Sph-1-P release in vitro, just as for the CXC chemokine measurement described above. However, the choice of EDTA or CTAD scarcely affected the plasma concentration of Sph-1-P; chelation of divalent cations with EDTA to inhibit coagulation was sufficient and CTAD was not necessary for the preparation of samples for Sph-1-P measurement, unlike for the measurement of the CXC chemokines. 22 This may be related to the difference in localization between Sph-1-P and the CXC chemokines in the platelets; Sph-1-P is a bioactive lipid basically derived from the membrane lipid bilayer, while the CXC chemokines are stored in and released from platelet-specific α-granules. 29
By employing the above-described Sph-1-P assay and plasma preparation methods, we found that while the plasma Sph-1-P concentration was not correlated with age, it was significantly higher in men (413.1 ± 52.0 nmol/L) than in women (352.4 ± 39.7 nmol/L). To the best of our knowledge, ours is the first study to report a difference in the plasma Sph-1-P concentration between the two sexes, which we believe was revealed only because of the reliable measurement method and suitable samples used by us. However, the more noteworthy finding of our study was the close correlation between the plasma Sph-1-P concentration and RBC-related parameters, and not platelet-related parameters. We had presupposed that the plasma Sph-1-P concentrations would be influenced by platelet-related parameters, because it is well established that Sph-1-P is abundantly stored in platelets and is released from the cells upon stimulation. 7
From a different viewpoint, however, this unexpected finding may not be all that surprising. Although we previously reported that the mol% of Sph-1-P/phospholipid value for platelets is over 10 times higher than that for RBCs, 30 the cell volume (reflecting the phospholipid amount) of RBCs is far greater than that of the platelets. Furthermore, it was found that sphingosine can be incorporated into RBCs and then converted to Sph-1-P and that a significant percentage of Sph-1-P formed inside RBCs may leak out even without stimulation of the cells. 31 Accordingly, it may not be so unexpected for RBCs to be an important source of plasma Sph-1-P. In support of this contention, it was very recently reported that RBCs store and release Sph-1-P in the blood. 32,33 Since the release of Sph-1-P from RBC is reportedly temperature-dependent, 33 the source of undesired Sph-1-P increase in plasma (Figure 2c) may be RBC. Furthermore, Pappu et al. reported that plasma Sph-1-P is mainly haematopoietic in origin, with RBCs being a major contributor, by using mice in which the two kinases that generate Sph-1-P were conditionally ablated. 34 Taken together, RBCs should be newly recognized as an important source of Sph-1-P in the blood, and the higher plasma Sph-1-P concentration observed in men (than in women) is likely to be due to the higher values of all the RBC-related parameters in men. Although DHSph-1-P was also detected in the plasma, there was no correlation between the plasma DHSph-1-P concentration and RBC-related parameters.
Examination of the correlations between the plasma Sph-1-P and the lipid profile revealed that the plasma Sph-1-P concentration was closely correlated with the serum total cholesterol, LDL-cholesterol (but not HDL-cholesterol) and apolipoprotein B. This was also an unexpected finding, since HDL has been reported as an important Sph-1-P carrier in the plasma; it has been suggested that the HDL-bound Sph-1-P may be responsible for the anti-atherosclerotic effects of HDL. 35–38 In addition, it was reported that the Sph-1-P concentration in HDL was much higher than that in apolipoprotein B-containing lipoprotein and lipoprotein-deficient plasma and that HDL-Sph-1-P was the major determinant of the plasma Sph-1-P concentration. 39 Although the reason for this discrepancy remains to be resolved in future investigations, analysis of the plasma Sph-1-P concentrations using a reliable assay method may unveil hitherto unknown significant roles of Sph-1-P in vivo. One of the limitations of our present study was that only healthy subjects were examined; evaluations in various patient populations including anaemia and hypercholesteraemia were to be expected.
Footnotes
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and the Japanese Society of Laboratory Medicine Fund for the Promotion of Scientific Research.