
Abstract
A pheochromocytoma is a rare catecholamine secreting neuroendocrine tumor, originating within the adrenal medulla. Most cases present with a hypertensive crisis and a classic triad of headaches, sweating, and palpitations. A positive prognosis is projected for patients after operative removal of the tumor. Early detection of this tumor is crucial as it can be fatal if left untreated. Clinical signs and symptoms, diagnostic criteria, and imaging appearances are described to assist in this case of a pheochromocytoma, detected in a pediatric patient. A 16-year-old female presented to the hospital with symptoms of chest pain, hypertensive crisis, and vision loss. A renal sonogram was performed and revealed a left sided adrenal mass. Correlating the sonogram with magnetic resonance imaging (MRI), tumor markers, and clinical presentation confirmed this pathology as a benign pheochromocytoma in a pediatric patient with hypertension. Medical and surgical intervention was provided to yeild a positive outcome for this patient.
Pheochromocytomas originates within the adrenal medulla and develops from chromaffin cells, produced by the adrenal gland. These tumors can induce numerous life-threatening symptoms and surgical intervention is typically needed as treatment in the patient’s plan of care. The estimated prevalence of pheochromocytomas is 1:2500 to 1:1650 with an incidence ranging from 1000 to 2000. 1 Of the incidence, 5% to 20% represent pediatric patients, and an additional 5% to 20% represent metastatic disease. 1 Pheochromocytomas can be an incidental finding on asymptomatic patients or may be detected due to a combination of symptoms. Hypertension, headaches, perspiration, palpitations, tremors, and facial pallor are the most common symptoms of having this tumor. 2 Pheochromocytomas can present as benign or malignant based on the Pheochromocytoma of the Adrenal gland Scaled Score (PASS). Further medical issues can arise if this tumor is left untreated. The description of this case study should help sonographers identify pheochromocytomas in a pediatric patient presenting with hypertension. The combination of tumor markers, magnetic resonance imaging (MRI), and sonography should inform sonographers on how to assist clinicians in forming a proper pheochromocytoma diagnosis, in a hypertensive pediatric patient.
Case Report
A 16-year-old female presented to the hospital with symptoms of chest pain, hypertensive crisis (blood pressure 240/30 mmHg), and vision loss due to a left retinal hemorrhage. She was admitted to the pediatric intensive care unit with no prior medical history, serious illnesses, or hospitalizations. A renal sonogram was ordered due to the hypertensive emergency. A sonogram was performed using a GE LOGIQ E10 (GE Medical, Waukesha, WI) and a C2-9 MHz transducer. Upon evaluation of the left upper quadrant, a round mass was found superior and medial to the left kidney and adjacent to the splenic hilum. This area measured 3.3 × 3.0 × 3.7 cm (see Figure 1). The sonographic appearance displayed a well-circumscribed hypervascular and hypoechoic mass with an echogenic center (see Figure 2). An MRI of the abdomen was then ordered and performed to support the suspected sonographic diagnosis of a pheochromocytoma, in the setting of hypertension. On the MRI images, the round lesion was identified at the level of the left adrenal gland and provided the signal characteristics that helped to make the diagnosis (see Figure 3). Lab testing revealed an elevated level of C-Reactive protein (CRP) of 3.3 mg/dL (normal < 0.9 mg/dL). The tumor markers metanephrine plasma and normetanephrine free plasma were evaluated. Metanephrine plasma measured a value of <75 pg/mL (normal 10–95 pg/mL) and normetanephrine free plasma measured 5900 pg/mL (normal 22–83 pg/mL).
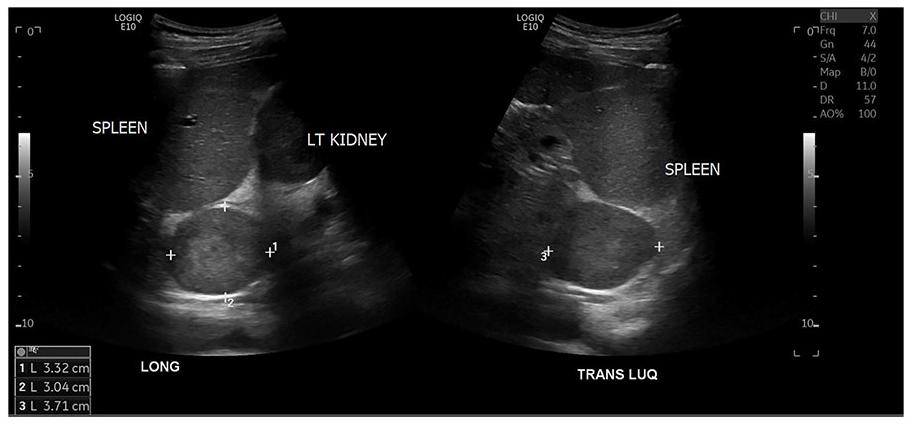
A pheochromocytoma that was measured in long and transverse imaging planes, on the dual sonogram.
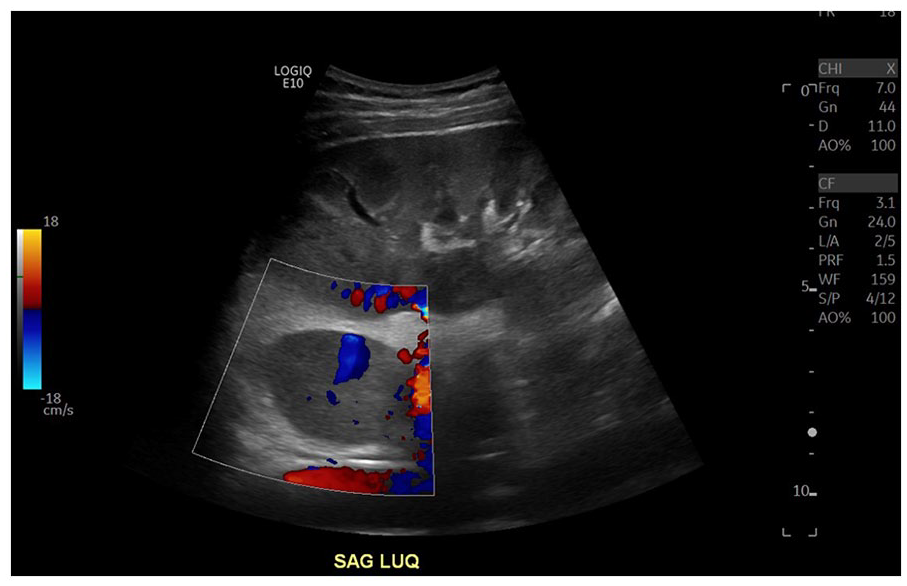
Color Doppler was used to evaluate this suspected hypervascular and hypoechoic left upper quadrant mass.
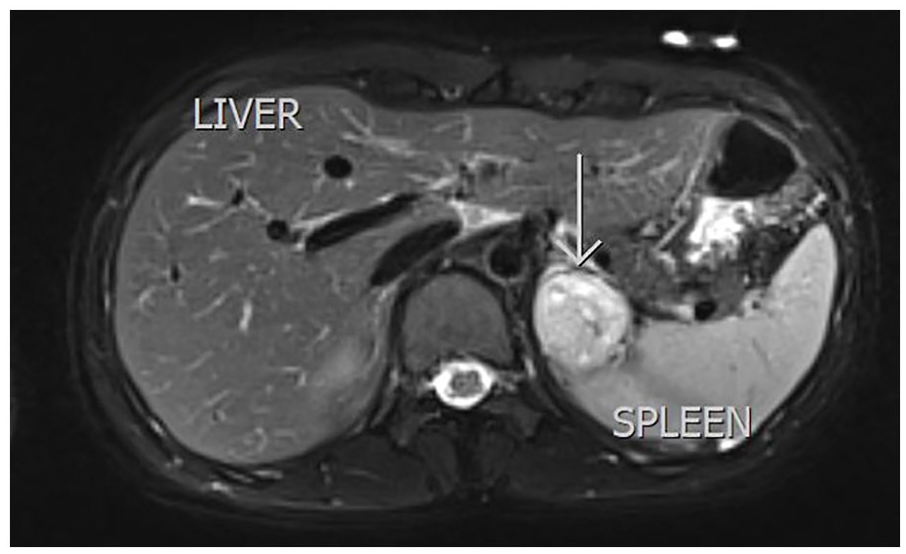
A magnetic resonance imaging slice was taken with a T2FS BLADE protocol. It demonstrates a heterogeneous T2 hyperintensity typical appearance of pheochromocytomas on MRI (marked with a white arrow).
A robotic left transabdominal lateral adrenalectomy was performed 2 weeks after the MRI examination. The patient was alpha blocked for 2 weeks preoperative to control hypertensive symptoms. A total left adrenalectomy was performed and identified a 4.2-cm mass arising from the left adrenal gland medulla. No periadrenal tissues were invaded by the tumor. The tumor consisted of areas of necrosis and vascular invasion. Rare mitotic figures (1 mitotic figure/10 high power fields) were also present with no atypical mitotic figures identified. There were no other pathological findings, therefore making the PASS score 3. Three days postoperative the CRP level was elevated to 4.8 mg/dL, then decreased to 0.3 mg/dL, 1 month postoperative. The normetanephrine free plasma was slightly elevated at 96 pg/mLp 2-months postoperative, then decreased to 48 pg/mL 5-months postoperative.
Discussion
Pheochromocytomas are rare catecholamine secreting neuroendocrine tumors originating within the adrenal medulla. These tumors can be categorized as either benign or malignant. When characterizing pheochromocytomas, the “10 percent rule” is applied. These rules state that these tumors are 10% extra-adrenal, 10% are bilateral, 10% malignant, 10% are found in asymptomatic patients, and 10% are familial. 3 Clinical symptoms are produced from secretion of excess catecholamines such as epinephrine, norepinephrine, and dopamine. 2 Clinical signs and symptoms associated with pheochromocytomas are nonspecific to the tumor. Hypertension with the classic triad of headaches, sweating, and palpitations are typically seen in patients suffering with this pathology. 3 Other symptoms consist of pallor, nausea, vomiting, constipation, weight loss, weakness, fever, pain, and anxiety. 2
Malignant hypertension, in the presence of a pheochromocytoma, can be linked to hypertensive retinopathy. 4 In this instance, the patient suffered from a retinal hemorrhage due to the malignant hypertension caused by the pheochromocytoma. Only 0.5% to 2% of hypertension in children are caused by pheochromocytomas. 2 Most pediatric patients with secondary hypertension are due to renovascular (12%–13%), renal parenchymal, and structural disease (78%), as well as endocrine, cardiac, and pulmonary causes. 5 Alpha blockers can be used to treat the hypertension preoperatively. Postoperatively, the excision of the mass will alleviate hypertensive symptoms. However, the patient will be monitored for hypertension for 1 year after diagnosis.
Diagnosis of this tumor can only be definitive through excision of the mass. Excision of the mass is the only form of treatment to completely diminish all symptoms. Malignancy is determined postoperatively while assessing the histology of the excised specimen. 6 The system of determining malignancy in a pheochromocytoma is called the PASS (Pheochromocytoma of the Adrenal gland Scaled Score) score. 7 This system consists of assigned points to grade the mass based on its macroscopic and microscopic characteristics. The points scale is out of 20 points. A PASS score less than or equal to three is treated as benign and has less malignant potential. A PASS score greater than or equal to four is treated as malignant and has more malignant potential. 7
Prognosis of pheochromocytomas differentiates between patients due to each individualized tumor. Certain factors such as age at diagnosis, male sex, large tumor size, dopamine hypersecretion, metastases, and declining surgical removal can cause rapid progression of the tumor and increase mortality. 8 An adrenalectomy carries a mortality rate of about 2% to 4%, with morbidity being as high as 40%. 4 Patients are monitored postoperatively for hypertension for several weeks. Some patients may require medication to resolve postoperative hypertension. However, this is only likely if the patient has residual lesions or metastatic disease. 5
Pheochromocytomas can be seen on computed tomography (CT), MRI, and with sonography. Discovering a pheochromocytoma solely through a sonogram is a unique finding, as the adrenal glands are normally not visualized in children and adults. The sonographic appearance is described as a well-circumscribed neoplasm originating in the adrenal medulla that varies from solid to complex. Anechoic areas with posterior enhancement may be seen in the setting of necrosis, hemorrhage, and liquefaction of the tumor. Sonographic evaluation can be beneficial in differentiating a mass between cystic and solid. However, the left adrenal gland can be difficult to evaluate on a sonogram due to the location of the stomach. The technical limitations of sonography when attempting to detect a pheochromocytoma require additional imaging, such as MRI to confirm a preoperative diagnosis.
MRI is the preferred imaging technique to diagnose a pheochromocytoma, in a pediatric patient. With MRI, these tumors present as hypointense signal on T1-weighted images and hyperintense signal on T2-weighted images. 2 About 30% of pheochromocytomas display a “light bulb sign,” which presents as a very hyperintense signal in the T2-weighted images. 2 Heterogeneous signals may indicate presence of bleeding, cystic degeneration, and calcifications. 2 Furthermore, the MRI images can help confirm the diagnosis and lead to treatment for the patient. A drawback to utilizing MRI, in this scenario, is that the patient had to be sedated to complete the examination. Differential diagnoses of an adrenal neoplasm on both sonography and MRI include adrenal adenoma, adrenocortical carcinoma, adrenal neuroblastoma, adrenal metastasis, and adrenal lymphoma. 3 The validity and reliability of this pathology on sonography and MRI both relies on the PASS score.
The gold standard to discovering pheochromocytomas prior to excision is biochemical lab work. The identification of catecholamine hypersecretion is used to measure plasma-free or urinary fractioned metanephrines. 1 The measurements of metanephrine, normetanephrine, methoxytyramine, and chromogranin A are utilized to determine the secretory profile of the tumor. The tumor marker of interest in this case is the normetanephrine free plasma as it was the highest in value in the patient’s lab work analysis. If these tumor markers are assessed when suspecting a pheochromocytoma in a pediatric patient, ultrasound could be the next step in identifying pathology. Imaging such as sonography, MRI, and CT can help locate the neoplasm and determine size and characteristics. Utilizing sonography as a baseline for pheochromocytoma screenings can significantly decrease the amount of radiation exposure to the patient.
Sonography is not the diagnostic gold standard when imaging an adrenal mass. However, sonographers should be aware of its appearance as it can easily be detected with sonography. These sonographic findings helped the patient avoid unnecessary radiation and expedite the care plan by finding the solution to their hypertensive crisis. Patients continue to be monitored for any signs of recurrence for several years after diagnosis. The prognosis of no recurrence is likely; however, some cases do have recurance. 9 A combination of lab work and imaging will be the focus of the care team in the following postoperative years.
Conclusion
Although pheochromocytomas are rare, it is important for sonographers to be able to identify them using sonographic appearance, signs, and symptoms. Sonographers should look for a well-circumscribed neoplasm varying from solid to a complex appearance, within the area of the right and left adrenal gland. In cases of pediatric secondary hypertension, sonographers must be aware of the possibility of a pheochromocytoma during a sonographic evaluation. Correlating the sonographic identification and classic triad of symptoms (headaches, sweating, and palpitations) can assist the clinical team in further patient management. This specific case study helped confirm a diagnosis of a symptomatic pediatric patient without the need for ionizing radiation, such as CT imaging. Using sonography as a baseline helps prevent unnecessary ionizing radiation to the pediatric patient and is ultimately putting the patient’s safety first. With imaging, tumor markers, and the PASS system, clinicians are placed in the most appropriate position to evaluate and diagnose a pheochromocytoma.
Footnotes
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Informed Consent
Informed consent was not sought for the present study because all case data was de-identified and/or aggregated and followed ethics committee or IRB guidelines (also referred to as the Honest Broker System).