
Abstract
A long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency constitutes a very rare autosomal recessive disease, affecting long fatty acids’ metabolism. In the prenatal period, it is difficult to suspect this diagnosis in the absence of family history, as there are no specific sonographic signs of this disease. A 32-year-old pregnant woman was receiving follow-up care, as she was a carrier for Duchenne muscular dystrophy (DMD). The fetus was female, and the pregnancy progressed normally until 29th week of gestation. At that time, the fetal bowel had noted dilatation with mild hyperechogenicity. Fetal surveillance was conducted with sequential sonograms. Labor was induced at 36 weeks, due to pre-eclampsia. The newborn presented with poor weight gain and hypoglycemic episodes. Neonatal screening revealed the diagnosis of an LCHAD deficiency. Prenatal diagnosis of LCHAD is usually performed by genetic testing when there is a family history. However, only a few sonographic signs have been described in association with this disease, such as fetal bowel hyperechogenicity or cardiomyopathy. This is the first case study to report a prenatal sonographic finding of bowel dilatation in a fetus with LCHAD deficiency, and one of the few to report fetal bowel hyperechogenicity.
Keywords
A long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency constitutes a very rare autosomal recessive disease with a worldwide birth prevalence estimated at 1/250 000.1–3 It affects long fatty acids’ metabolism, due to an enzymatic failure in the third step of the mitochondrial beta-oxidation. 4 Consequently, there is impaired production of energy and accumulation of acylcarnitine intermediates. 2
The clinical presentation of an LCHAD deficiency can range from non-specific symptoms, like hypoglycemic events and poor weight gain, to more severe consequences. 4 The most severe forms may present with a rapidly progressive cardiomyopathy in the neonatal period. 5 Later, the infants may develop hypoketotic hypoglycemia with metabolic acidosis, resulting in liver dysfunction, rhabdomyolysis, seizures, or death. 4 Skeletal myopathy, peripheral neuropathy, and pigmentary retinopathy may be observed in surviving individuals.4,5
The prenatal diagnosis may be easily done when there is family history, by searching for known familial mutation or measuring enzyme activity in cultured cells obtained from the amniotic fluid or chorionic villi samples. 2 However, in the absence of family history, it is very difficult to diagnose LCHAD deficiency. In the fetal period, the occurrence of cardiomyopathy with pleural effusion and/or hydrops, intrauterine growth restriction, and increased bowel echogenicity have been reported.3,6,7 Furthermore, LCHAD deficiency in the fetus has been associated with pregnancy complications, specifically hypertensive disorders. 8
This case report adds another sonographic finding to those already reported that may be associated to LCHAD deficiency.
Case Report
A 32-year-old Caucasian pregnant woman, G3P0, in the tenth gestational week, was referred to a tertiary prenatal diagnosis center due to a 50% dystrophinopathy recurrence risk in the case of male fetus. The patient was a carrier (heterozygous) for a pathogenic variant in the dystrophin gene (Duchenne muscular dystrophy [DMD]), which had been identified by Multiplex Ligation-dependent Probe Amplification, due to an affected male sibling.
In the medical history of the patient, there was idiopathic chronic hypertension and a combined cobalamin and iron deficiency anemia. Regarding obstetric history, it included one intrauterine fetal demise of unknown cause at 25 weeks of gestation, and one medical termination of pregnancy. In those cases, both male fetuses suffered from a genetically confirmed DMD.
In the current pregnancy, the patient underwent an early non-invasive prenatal testing (NIPT), with the main objective to determine the fetal gender, due to the recurrence risk for DMD’s pathogenic variant. The sex-determining region Y protein was absent, and a female fetus was assumed. In addition, the NIPT revealed a low risk for chromosomal abnormalities. The sonograms performed at 10, 12, 18, and 22 weeks were all normal, and confirmed the female fetal gender. At 29-week gestation, an axial sonographic view of fetal abdomen revealed fetal bowel dilatation (FBD), with a maximum diameter of 10.8 mm (normal range: < 7 mm) (see Figure 1), and an estimated fetal weight (EFW) in the 20th percentile. Furthermore, bowel echogenicity appeared to be increased (see Figure 2), with no other detectable sonographic abnormalities.
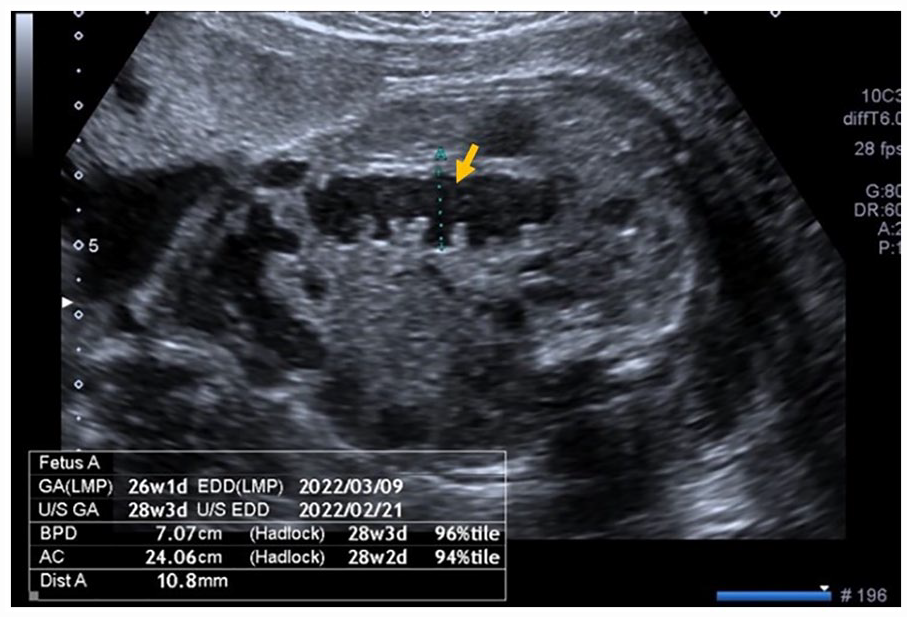
An axial sonographic view of a 29-week fetal abdomen is provided and demonstrates dilated loops of bowel that measure 10.8 mm (see arrow).
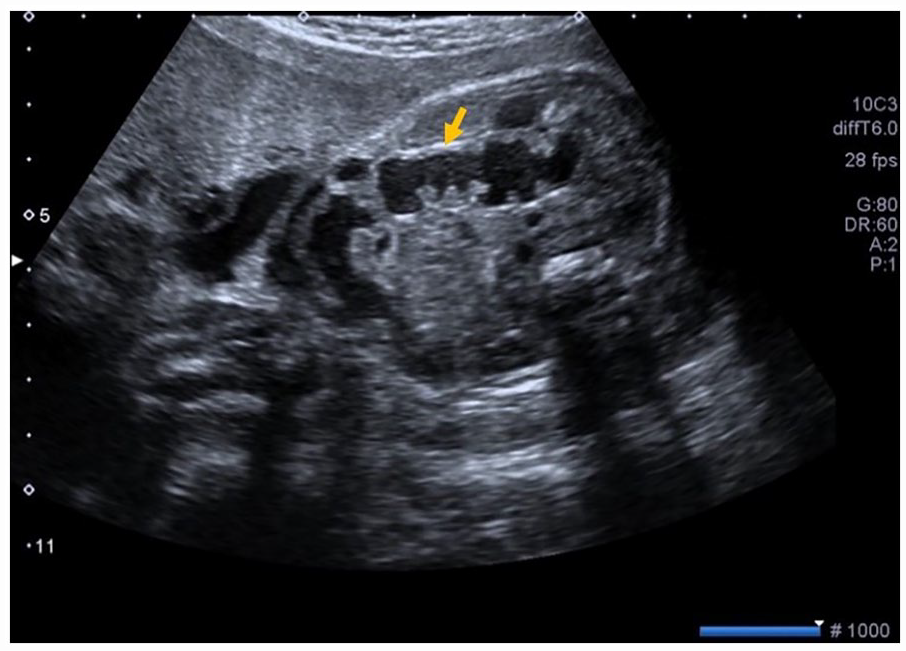
An axial sonographic view of a 29-week fetal abdomen is provided and demonstrates increased bowel echogenicity (note the arrow pointing to the intestine wall with similar brightness compared with fetal bone).
Fetal growth and bowel dilatation were monitored every 2 weeks. At the 31st, 33rd, and 35th weeks, the EFW decreased to the 5th–10th percentile, with persistence of bowel dilatation (see Figure 3). The Doppler evaluation of the umbilical artery and the middle cerebral artery, as well as the volume of amniotic fluid, remained normal during the pregnancy.
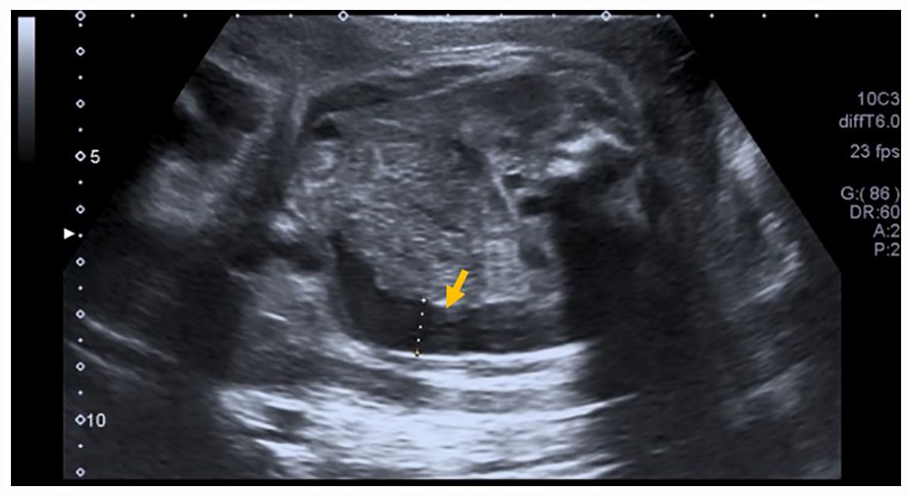
An axial sonographic view of a 31-week fetal abdomen that demonstrates dilated loops of bowel (see arrow).
At 36-week gestation, the patient presented with increasing blood pressure and positive urinary protein to creatinine ratio, consistent with pre-eclampsia. Due to this diagnosis, the patient was admitted for induction of labor that ended in a eutocic delivery. The female newborn weighed 2210 g (17th percentile) and the Apgar Index was 9/10/10. After delivery, there were clinical and analytic improvements, which allowed hospital discharge on the second postpartum day. A syndactyly of the fourth and fifth left toes was the only newborn physical anomaly identified.
The newborn was re-admitted to the hospital during the neonatal period, due to poor weight gain and hypoglycemic episodes. Infectious causes and cardiopathy were excluded. The neonatal screening test detected LCHAD deficiency. The profiles of urinary long-chain fatty acids and acylcarnitines obtained from dried blood spots (Guthrie card) confirmed the diagnosis. Molecular analysis of HADHA gene identified the c.1528G > C (p.E510Q) pathogenic variant in the newborn, in homozygous state, confirmed after parent’s analysis. Milk supplementation with low lipid plus high content of medium-chain triglycerides was started, followed by progressive weight recovery and hospital discharge.
Discussion
The determination of fetal sex is mandatory when a pregnant woman is a carrier of DMD. This genetic disorder linked to X chromosome is a severe condition that affects male descendants of female carriers with a 50% chance.2,3 The early fetal gender determination by NIPT allowed to waive an invasive technique and its inherent, albeit small, risk of miscarriage. 9 An earlier intervention decreases psychological distress for the patient and allows a less complex medical termination of pregnancy. 9
The diagnosis of LCHAD deficiency, although unexpected due to its low incidence, was consistent with many findings in this case, in particular the hypertensive complication that occurred at the end of pregnancy. 8 In fact, LCHAD deficiency has been connected to various pregnancy complications, such as severe pre-eclampsia, hemolysis, elevated liver enzymes, low Platelet (HELLP) syndrome, acute fatty liver of pregnancy, fetal growth restriction, or even intrauterine fetal demise. 8 The patient developed a late pre-eclampsia without severe features, but the patient also had chronic hypertension, thus providing a probable multifactorial etiology for this pregnancy hypertensive disorder. Furthermore, it was noticed a decreased in EFW percentile during the follow-up, which is consistent with LCHAD deficiency. Regarding the sonograms, this fetus presented two different findings: hyperechogenic bowel and bowel dilatation. The hyperechogenic bowel has been defined as bowel of similar or greater echogenicity than surrounding bone. 10 FBD is usually determined by the cut-off of > 7 mm in diameter of fluid-filled intestinal loops on sonography.8,11 Augmented bowel echogenicity has been linked to LCHAD deficiency at least in two fetuses. 7 Lee et al. described a case of a 23-year-old woman who had two children with a confirmed diagnosis of LCHAD, both with identification of hyperechogenic bowel in the prenatal period. 7
FBD is more commonly detected during the second and third trimesters of pregnancy, and there is a lack of data regarding the prognosis of its late-appearance.10,11 It can be a sign of mechanical or functional obstruction. 12 Among recognized etiologies are bowel atresia or stenosis, malrotation with volvulus, meconium ileus, total colonic aganglionosis or meconium plug syndrome. 12 Although it can persist on sequential sonograms without clinical significance in neonatal period, it can also be considered an indirect sign of adverse outcomes.8,11 In this case, an obstructive cause was not possible to exclude, but the stomach was not enlarged and there was no polyhydramnios (present in more proximal obstructions). 12
In LCHAD deficiency, the pathophysiological mechanism involved in the multiple organ expression is unknown. 7 Tercanli et al. reported one case of fetal cardiomyopathy diagnosed at 31 weeks of gestation, in a fetus with LCHAD deficiency, probably linked to muscular weakness or myopathy, frequent in cases of mitochondrial protein deficiencies. 3 Previous studies involving human embryos and fetal tissue have proved a multiorgan LCHAD expression, including in the gut. 6 In consideration of this, it is possible that the sonographic abnormalities of this fetus could also be explained by LCHAD deficiency. 7
The treatment of individuals affected by LCHAD deficiency involves a restrictive low-fat diet, through replacing long-chain fatty acids with medium chain ones, avoiding long fasting periods or energy consumption activities, like physical exercise or exposure to harsh environmental conditions. 1 However, even with dietary support, lifelong sequels may occur.1,4
Conclusion
Based on a limited review of the literature, this may be the first case reporting sonographic FBD in a fetus with LCHAD deficiency, and one of the few reporting increased bowel echogenicity. Although FBD is a nonspecific sign, it can increase clinical suspicion of LCHAD deficiency, and parental counseling and eventual transfer to a specialized tertiary center should be considered.
Finally, in the presence of family history of LCHAD deficiency and when a prenatal diagnosis is done, it is important for the sonographer to know which are the most common prenatal sonographic findings in this disease, so as not to increase anxiety in relation to other pathologies.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval was not sought for the present study because it is not required for case reports or case series.
Informed Consent
Informed consent was not sought for the present study because all case data were de-identified and/or aggregated and followed ethics committee or IRB guidelines (also referred to as the Honest Broker System).