
Abstract
Mayer-Rokitansky-Küster-Hauser syndrome is a disorder affecting the female reproductive system, which can result in an absent uterus and vagina or a partially absent vagina with a normal uterus. While this female reproductive disorder can be difficult to diagnose, transabdominal sonography is often the initial imaging modality used to evaluate the female pelvis in this patient population as many females are not diagnosed with the disorder until primary amenorrhea is noted around puberty. This case study presents a young female who is diagnosed with an absent uterus and vagina but normal ovaries and discusses the role that sonography plays in diagnosing Mayer-Rokitansky- Küster-Hauser syndrome.
Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome affects the female reproductive tract and has an incidence of approximately 1 in 5000 newborn females.1,2 MRKH syndrome is a congenital disorder characterized by uterovaginal atresia in a female who is otherwise phenotypically normal, having a 46,XX karyotype. 2 In diagnosed cases of MRKH syndrome, there is a related 15% to 40% incidence of urologic abnormalities,3,4 and a renal sonogram is typically ordered to assess for abnormally formed or positioned kidneys or for unilateral renal agenesis. 5 In 12% to 50% of MRKH syndrome cases, skeletal anomalies such as the absence or fusion of the vertebrae also are demonstrated.5–8 This suggests an association with Klippel-Feil syndrome (KFS), a rare skeletal disorder primarily characterized by abnormal union or fusion of two or more bones of the spinal column within the cervical vertebrae.3,9,10 The most common presentation of MRKH syndrome demonstrates a completely absent uterus and vagina; a hypoplastic vagina with a normal uterus is seen less often.1,5,7,8,11 Ovaries are typically normal in appearance and function, with no sign of androgen excess in individuals affected with MRKH syndrome. 11
In pediatric cases where MRKH syndrome is suspected, transabdominal pelvic sonography is the best initial imaging modality to evaluate the female pelvis. Transvaginal sonography is typically contraindicated in these cases, as this examination is generally done only for sexually active patients. Magnetic resonance imaging (MRI) may be used as a complement to sonography; hysterosalpingography is not useful in the evaluation of vaginal agenesis and hypoplasia.3,12,13 Because sonography can effectively evaluate the uterus, cervix, vagina, ovaries, and adnexa, it is the primary imaging method used to detect and diagnose MRKH in young female patients, as demonstrated in this case.
Case Report
A 17-year-old female presented to the ultrasound department for a transabdominal sonogram because of primary amenorrhea. The patient had normal secondary sexual development. She stated that she had never started menstruating but had developed breasts and pubic hair approximately three years ago. The patient had a history of and showed normal growth, measuring 162.6 cm in height and weighing 52.8 kg at the time of her examination.
A transabdominal sonogram was done using a GE Logiq E9 ultrasound machine (GE Healthcare, Milwaukee, Wisconsin) with a curved array C1-5 transducer. The patient’s bladder was fully distended, which allowed for optimal imaging of pelvic structures. Both ovaries were sonographically visualized during the initial overview of the patient’s pelvis. The sonographic protocol began at the midline of the pelvis in the sagittal plane. Midline, posterior to the urinary bladder, no uterus was visualized. A small hypoechoic structure was seen at the midline pelvis, in both sagittal and transverse planes, measuring 2.7 × 0.7 × 1.7 cm, possibly representing a hypoplastic uterus (Figures 1 and 2). To the right and left of midline, no apparent uterus was visualized (Figures 3 and 4).
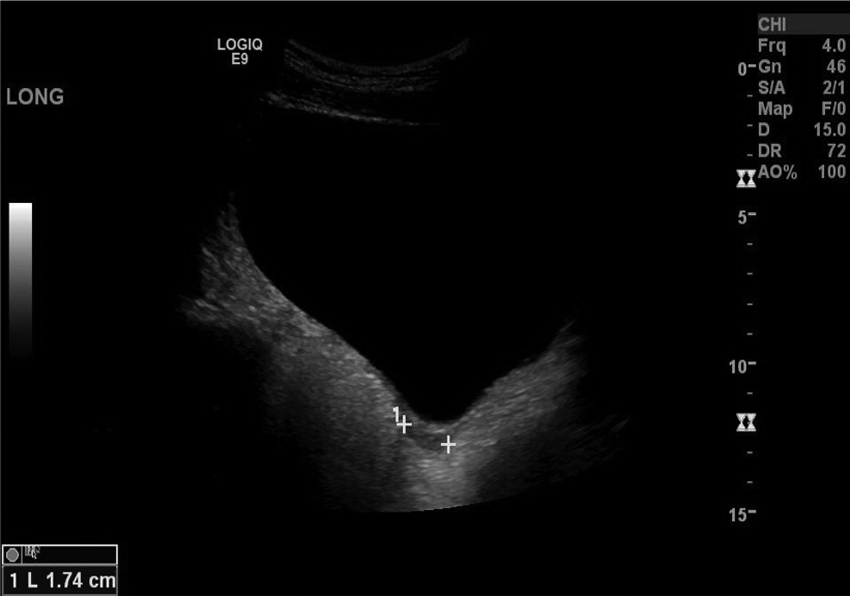
Transabdominal sonogram showing a small hypoechoic area located posterior to the bladder in the sagittal plane, possibly representing a hypoplastic uterus.
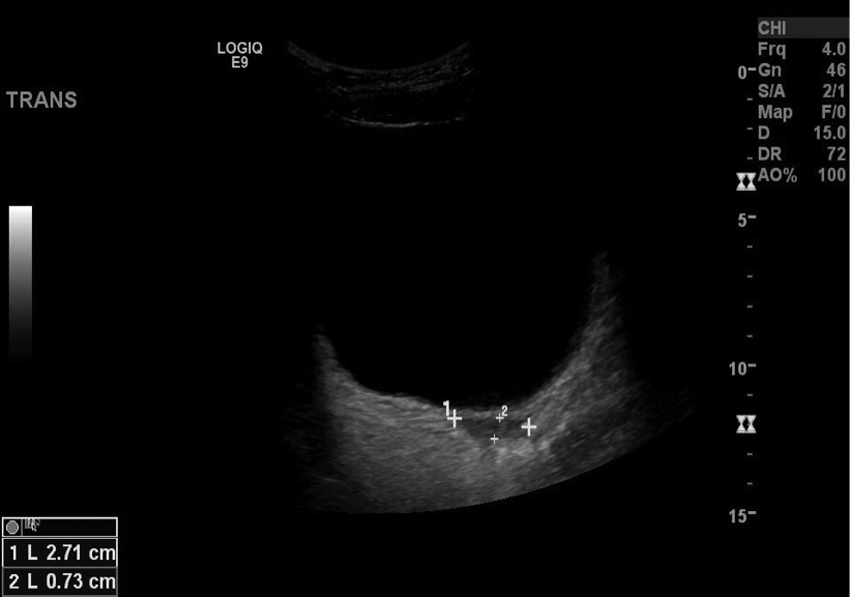
Transabdominal sonogram showing this same small hypoechoic area located posterior to the bladder in the transverse plane, possibly representing a hypoplastic uterus.
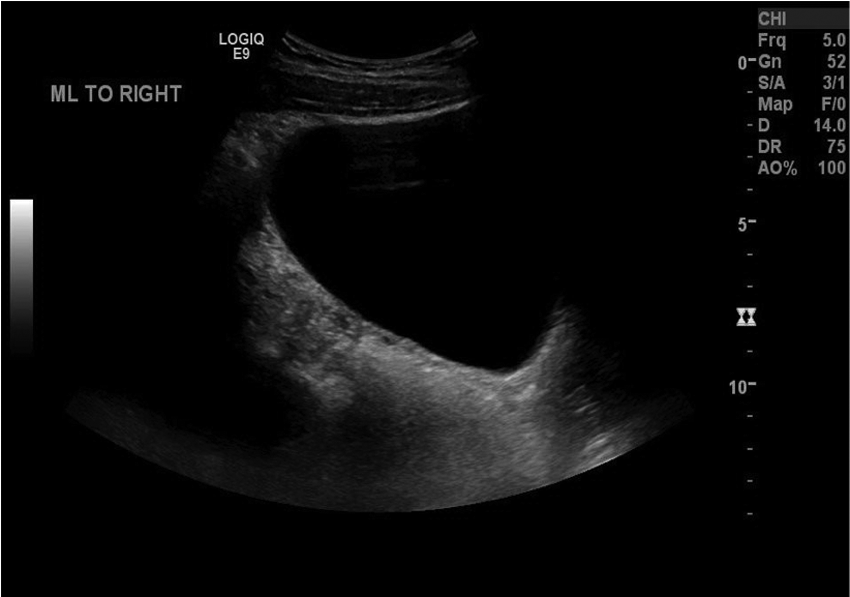
Transabdominal sonogram of the pelvis to the right of midline in the sagittal plane, showing the absence of a normal uterus.
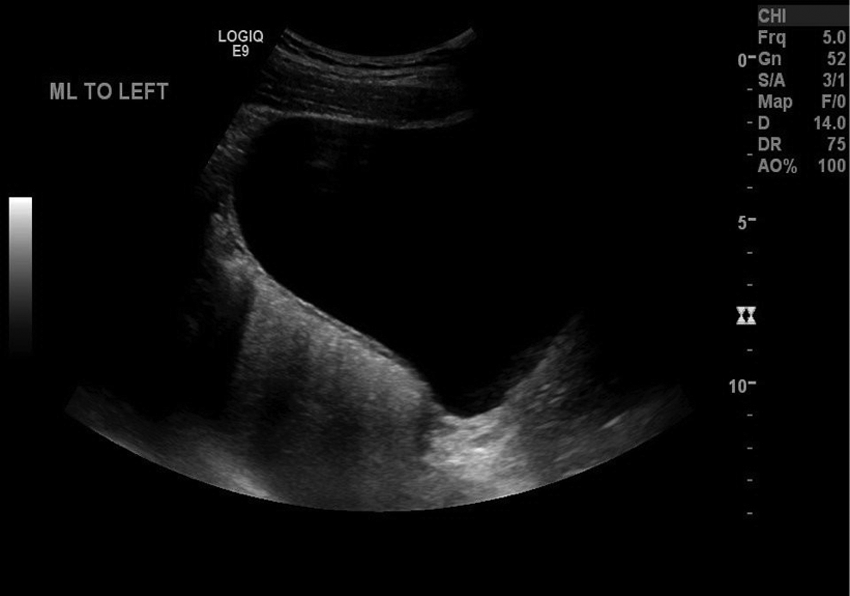
Transabdominal sonogram of the pelvis to the left of midline in the sagittal plane, showing the absence of a normal uterus.
The right and left ovaries were visualized, measuring 3.9 × 1.6 × 1.5 cm and 3.8 × 2.4 × 3.2 cm, respectively, and showed satisfactory blood flow when evaluated with pulse-wave Doppler (Figures 5–10). The left ovary was noted to contain a simple anechoic cyst that measured 1.6 × 2.6 × 2.4 cm (Figure 11). Both ovaries appeared sonographically normal.
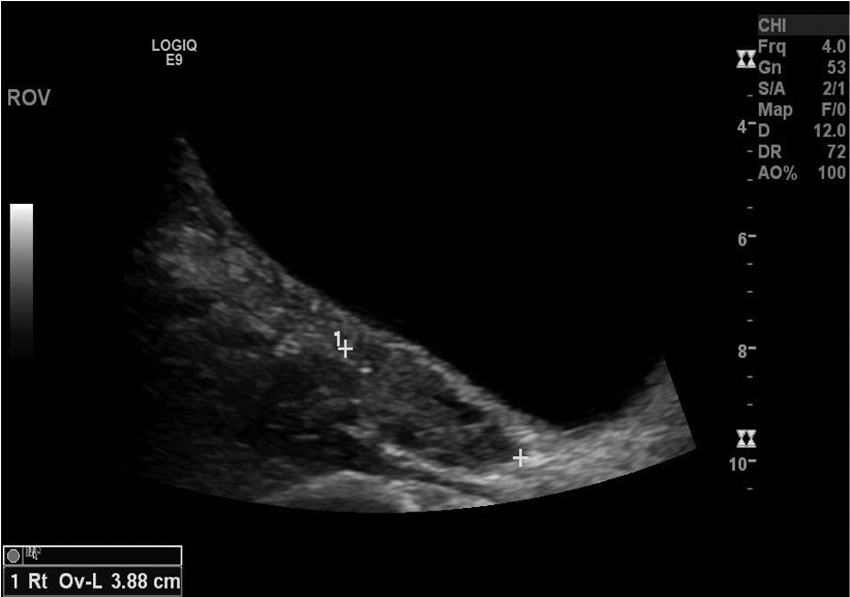
Transabdominal sonogram showing the right ovary in the sagittal plane.
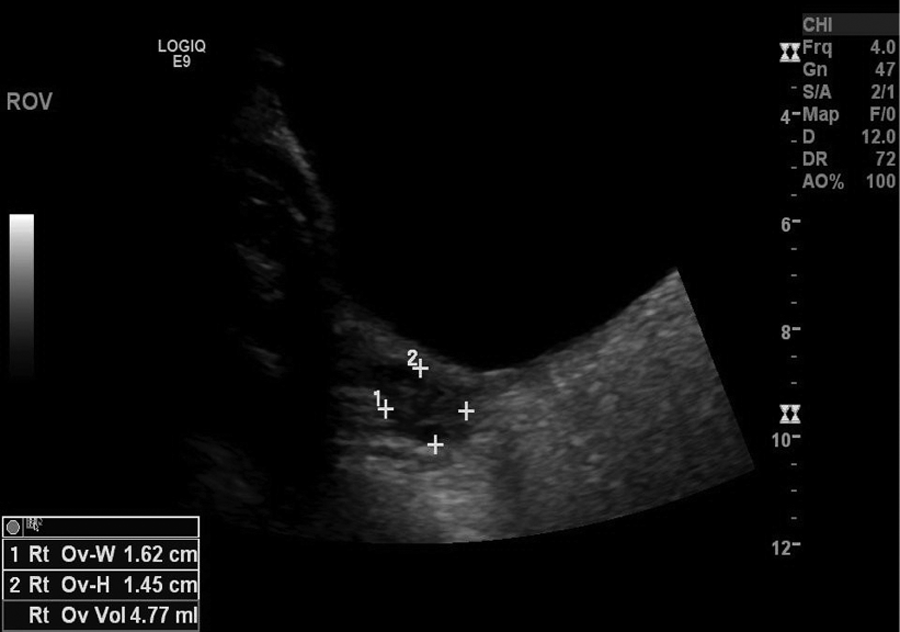
Transabdominal sonogram showing the right ovary in the transverse plane.
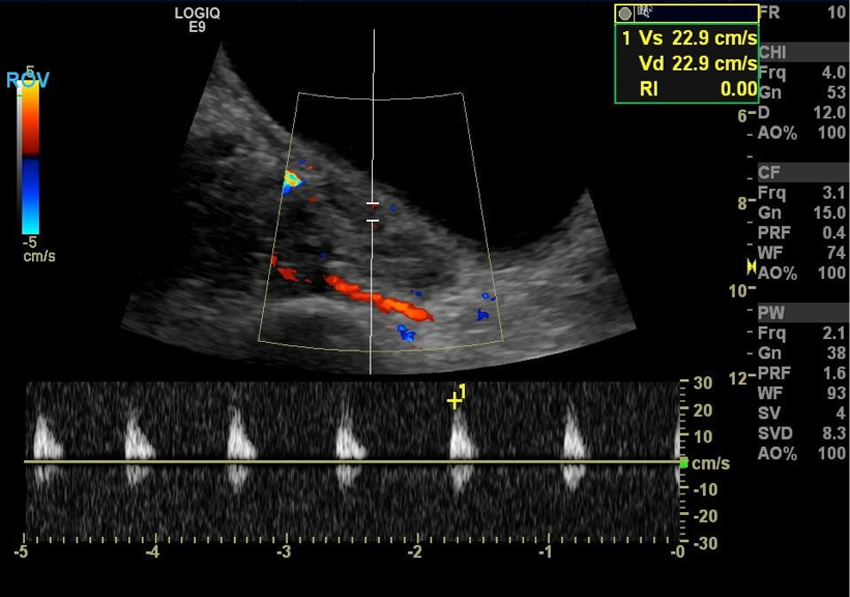
Transabdominal sonogram using color and spectral Doppler showing the detection of blood flow within the right ovary.
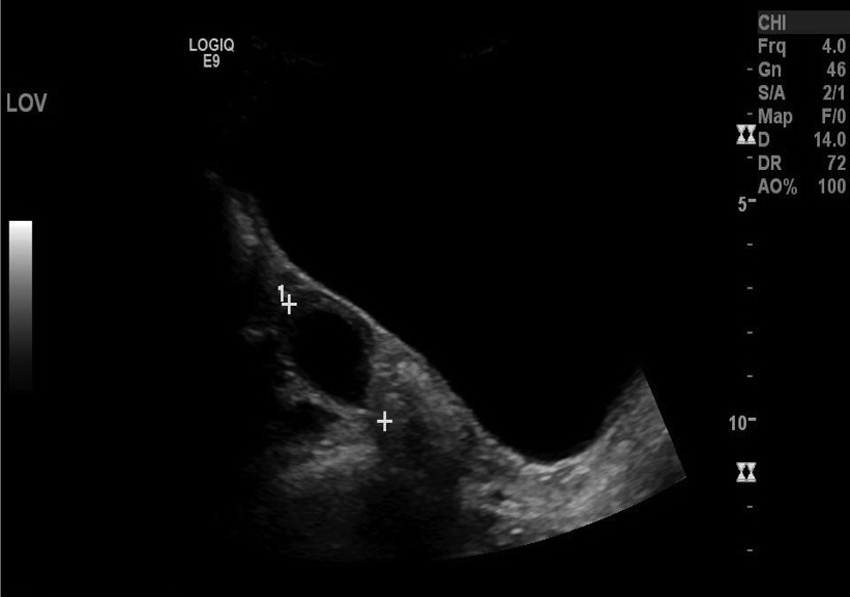
Transabdominal sonogram showing the left ovary in the sagittal plane, with incidental note made of a small cyst.
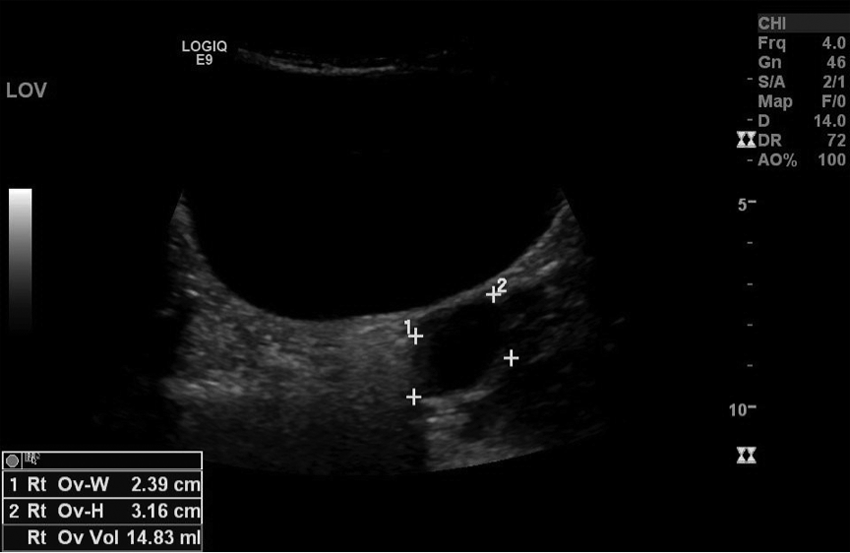
Transabdominal sonogram showing the left ovary in the transverse plane, with incidental note made of a small cyst.
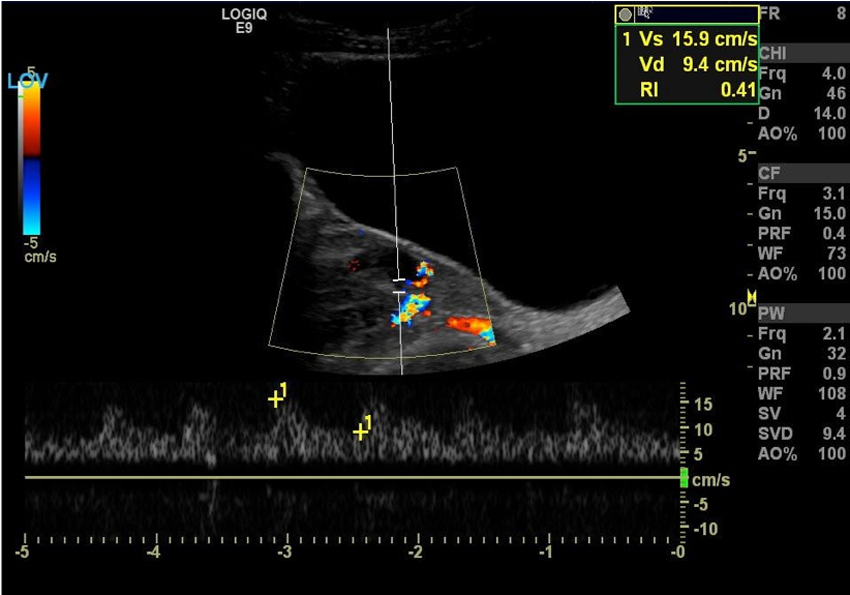
Transabdominal sonogram using color and spectral Doppler showing the detection of blood flow within the left ovary.
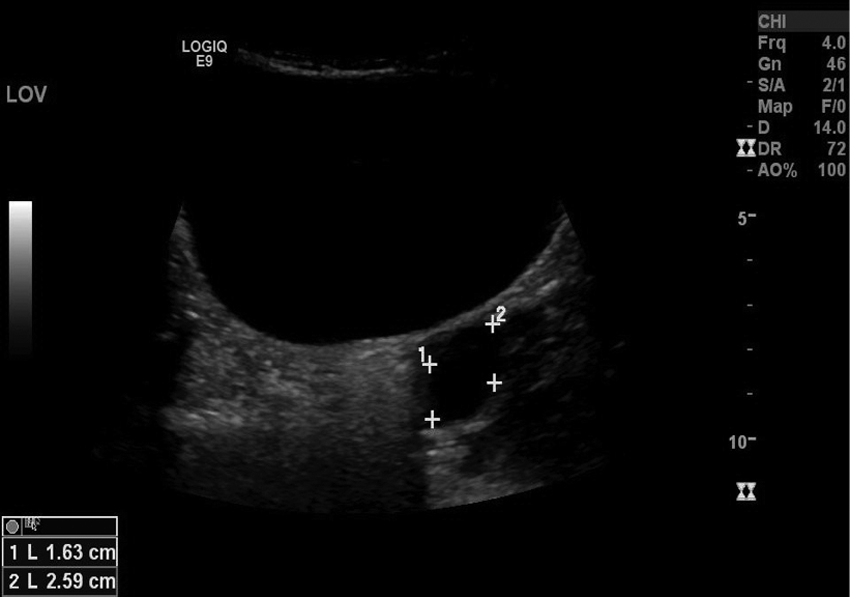
Transabdominal sonogram showing the left ovary in the transverse plane, with measurements of the left ovarian cyst.
The pelvis was then evaluated in the transverse plane, scanning from inferior to superior. The area where the uterus would typically be located was documented; however, no uterus was visualized (Figure 12). More superior to where the vagina should have been visualized, the small hypoechoic area was again seen. While this area could possibly be representative of a hypoplastic uterus, no normal-sized uterus was demonstrated sonographically.
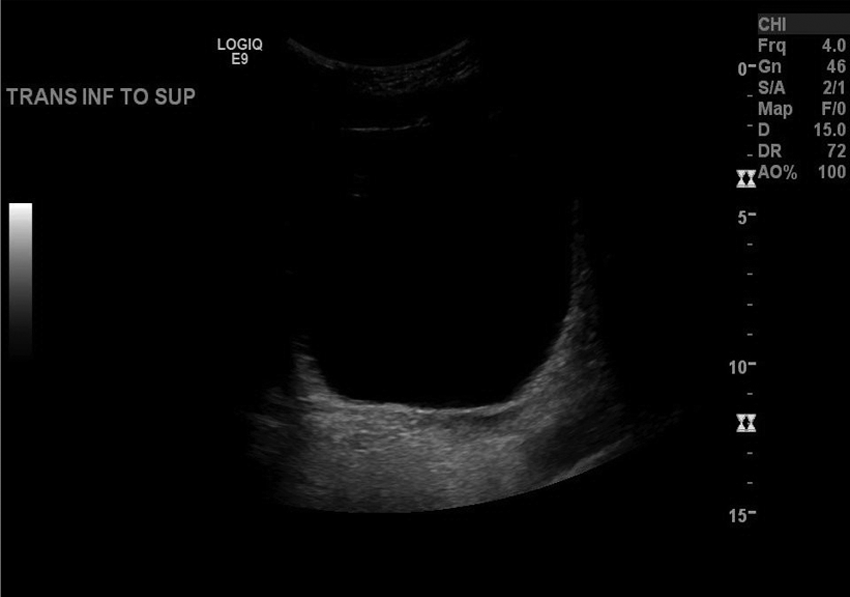
Transabdominal sonogram showing the pelvis posterior to the bladder in the transverse plane, with no uterus seen.
On the basis of the sonographic findings, the radiologist diagnosed the patient with possible hypoplastic uterus versus MRKH syndrome. The patient was then seen by a pediatric gynecologic specialist, who told her that she likely had MRKH syndrome. During the physical examination by the physician, an attempt to pass a Q-tip into a vaginal opening was unsuccessful and was met with immediate resistance. With the likely diagnosis of MRKH syndrome, the Müllerian anomaly warranted further sonography and testing to be performed.
A renal sonogram was ordered, as well as karyotype testing and follicle-stimulating hormone (FSH) and estradiol level measurements. Karyotype testing showed a normal female karyotype, 46XX. FSH was determined to be 8.2 mIU/mL, and estradiol was 149 pg/mL, both within normal limits. (Normal FSH levels for young women during puberty are 0.3–10.0 mIU/mL, and normal estradiol levels for premenopausal women are 30–400 pg/mL.14,15) The renal sonogram showed both the right and left kidneys in normal position and orientation, along with normal length measurements. The right kidney measured 10.4 cm in length (Figure 13), and the left kidney measured 11.6 cm in length (Figure 14).
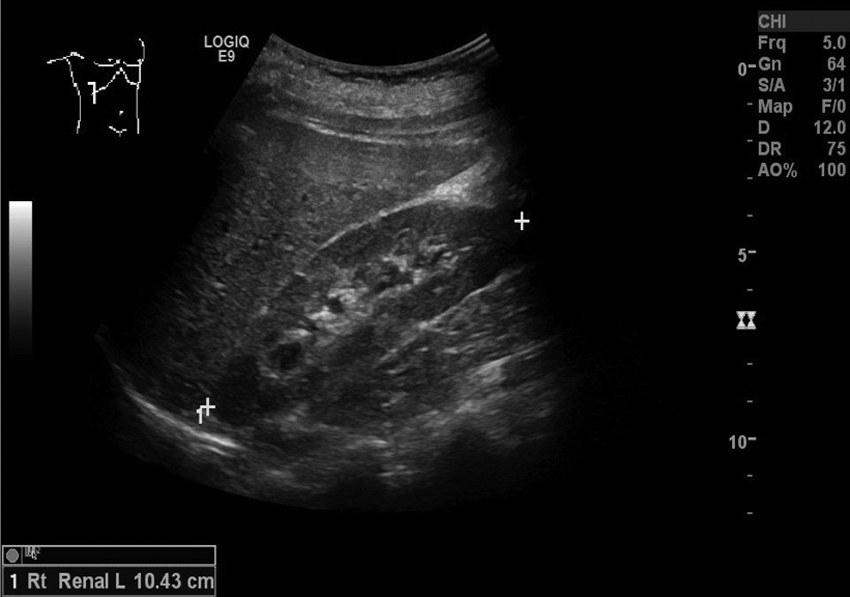
Sonogram showing a normal right kidney in the sagittal plane, measuring 10.4 cm in length.
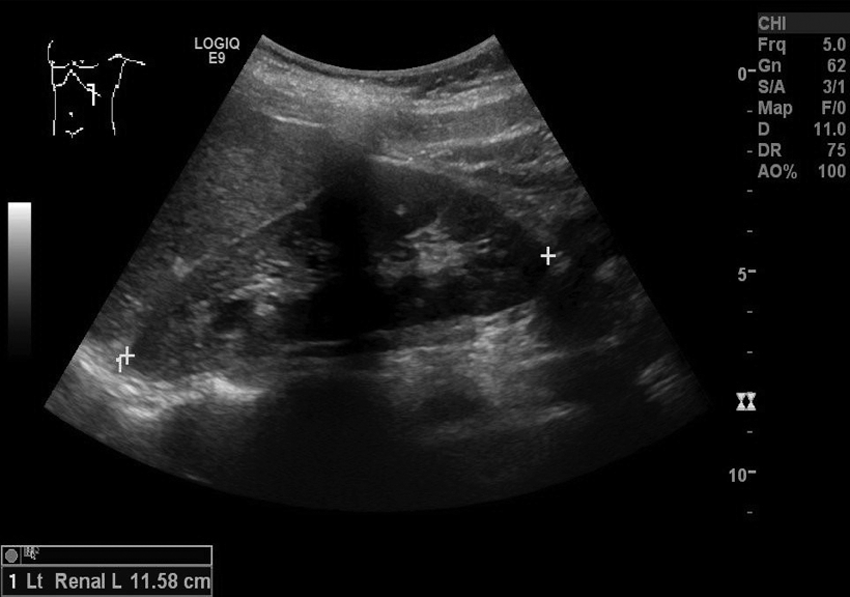
Sonogram showing a normal left kidney in the sagittal plane, measuring 11.6 cm in length.
On the basis of the findings of sonographic imaging and further testing, the physician confirmed the diagnosis of MRKH syndrome. Due to the lack of a functioning vaginal opening, a treatment option of vaginal dilation therapy under anesthesia was suggested.
Discussion
Sonographic imaging was the initial imaging modality used to determine the possible cause for this patient’s primary amenorrhea. Because of the patient’s age, only transabdominal pelvic sonography was used to evaluate the patient’s female reproductive organs, which demonstrated an absent uterus, cervix, and vagina, with normal right and left ovaries, consistent with MRKH syndrome.
MRKH syndrome is a condition occurring in approximately one in 5000 female newborns. 2 The 46,XX karyotype is what differentiates MRKH syndrome from other genital tract development defects such as Turner syndrome (45,0X) and androgen insensitivity syndrome (46,XY).11,16,17 In addition to having abnormalities of the reproductive tract, approximately 30% of affected individuals also will have renal abnormalities.5,8,11,18 The most common of these abnormalities is the absence of one kidney (unilateral renal agenesis) or the abnormal location of one or both kidneys. The kidneys may also be fused together at the inferior poles and sit midline in the abdominal cavity, a condition known as horseshoe kidneys. Approximately 12% of females with MRKH syndrome also have skeletal abnormalities usually affecting the spine, ribs, or limbs. A third common finding in patients with MRKH syndrome is auditory abnormalities.11,18
The exact cause of MRKH syndrome is unknown. Initially, MRKH syndrome was thought to occur randomly secondary to the involvement of nongenetic or environmental factors, but no correlation between environmental factors and MKRH syndrome has been established.2,5,11 Increasing evidence suggests that MRKH syndrome is a genetic disorder. It has been proposed that in familial cases, the syndrome is inherited as an autosomal dominant trait. However, some individuals who inherit the gene for a dominant disorder, such as MRKH syndrome, will not be affected by the disorder. 5
Most patients with MRKH syndrome have complete arrestment of the paramesonephric ducts, which yields a completely absent uterus and vagina. 3 Less commonly seen is a normal uterus with a small vaginal pouch, which occurs when there is partial arrestment of the paramesonephric ducts. 3 External genitalia appear normal, which typically means that MRKH syndrome will not be diagnosed until puberty.4,8,17 Diagnosis usually occurs around age 15 years, when it is noted that menses has not begun. Clinical signs that present at puberty are amenorrhea and possible cyclic pelvic pain, although MRKH syndrome can have widely varying signs and symptoms among affected individuals.3,5 The most common sign of MRKH syndrome, and often the first clinical symptom, is amenorrhea. Some affected individuals may present earlier with renal or skeletal abnormalities, as well as hearing loss, which warrants further investigation.
A similar case of MRKH syndrome has been reported in which a female teenage patient presented with primary amenorrhea and stated that she had become sexually active at 16 years of age. 17 The patient noted otherwise normal changes associated with puberty around age 13, including normal breast development, average height and weight, and pubic and axillary hair growth. A hormone profile on the patient, including FSH, estradiol, and 17-hydroxyporgesterone levels, was normal. Sonography confirmed the diagnosis of MRKH, showing a blind vagina and poorly formed uterus, with both ovaries appearing normal in size and appearance. Sonography was also able to diagnose a single left pelvic kidney. Genetic testing confirmed a 46,XX karyotype. This combination of findings led to the diagnosis of MRKH syndrome, and this previously reported case has striking similarities to the case study presented here.
The prognosis for individuals affected with MRKH syndrome is good. While individuals with MRKH do not have a vagina and typically do not have a uterus, both nonsurgical and surgical methods can be used to achieve the formation of a vagina.19–25 Nonsurgical treatment involves the use of graduated dilators to progressively widen an opening, creating a neovagina, a treatment less invasive than surgery. This formation of a vaginal opening will allow for sexual activity, but the process can take several months and sometimes years before a functioning vagina is formed.24,25 One form of surgical treatment that has been shown to be successful involves using the distal sigmoid colon to create a vagina. 26 Concurrent psychologic counseling and follow-up for these young female patients are also a primary aspect of management.27,28
Pregnancy in those with MRKH depends on the degree of hypoplasia of the uterus. Since the ovaries are normal, a pregnancy can be successful if enough functional endometrial tissue is present. In most cases, the uterus will be severely hypoplastic, so a pregnancy will not be achieved. However, individuals with MRKH syndrome are able to reproduce biological offspring since their ovaries are functional.3,11,29
The goal of a pelvic sonogram in cases where a patient is experiencing primary amenorrhea is to sonographically demonstrate the presence of the uterus, cervix, vagina, adnexa, and ovaries. Typically, an absent uterus will not be the cause of amenorrhea; however, it is important not to completely rule out the possibility of MRKH syndrome when examining pediatric patients. It is important that the sonographer ensure that the patient has a fully distended bladder in order to adequately evaluate the pelvic anatomy. The sonographer must also have thorough knowledge of the normal appearance of the female pediatric pelvis to determine if pelvic abnormalities are present. A sonographer’s understanding of the examination findings, as well as the patient’s medical history and clinical presentation, is what ultimately leads to an accurate diagnosis.
Conclusion
MRKH is a clinical finding that should be considered when young females experiencing primary amenorrhea have had secondary sexual development. This clinical information can be paired with the absence of the uterus and vagina upon sonographic examination to reach a definitive diagnosis. Detection of this syndrome can aid in determining a treatment plan for the patient and whether or not that plan will be surgical or nonsurgical. Because the treatment will affect future sexual function, it is important that MRKH be detected as early as possible. The earlier MRKH is diagnosed, the sooner treatment can begin and sexual function can be corrected.
Footnotes
Acknowledgements
We thank Lindsey Tegeler, RDMS, RVT, and the University of Missouri Diagnostic Medical Ultrasound staff for their assistance, leadership, and guidance.
Declaration of Conflicting Interests
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.