
Abstract
Solid-pseudopapillary tumor (SPT) of the pancreas is an extremely rare neoplasm in children, with an unknown origin. It accounts for 8% to 12% of primary pancreatic tumors and predominantly affects young females (91%), with very few cases reported in males. Malignant potential is minimal, particularly in adolescents. We report a 12-year-old child presenting with abdominal distension, nausea, vomiting, appetite loss, weight loss, pruritus, jaundice, dark urine, and back pain. Examination revealed a distended abdomen moving with respiration and a firm, tender 15 cm × 20 cm mass in the right upper quadrant. CT imaging demonstrated a large heterogeneously enhancing mass arising from the pancreatic head, consistent with SPT. Surgical therapy remains the treatment of choice, with partial duodenopancreatectomy or distal pancreatectomy (spleen-preserving when possible) recommended for complete excision. Although the body and tail are more commonly affected, complete resection (R0) offers excellent prognosis, even with local invasion or metastasis.
Introduction
In children, solid-pseudo papillary tumor of the pancreas (SPT), also referred to as solid pseudopapillary epithelial neoplasm (SPEN)is an extremely unusual tumor. Its origin is still unknown. Ninety-one percent of cases are in young females and has a minimal chance for malignancy, particularly adolescent girls. There have only been a handful documented examples of male SPT patients to date. A rare primary neoplasm of the pancreas, solid-pseudo papillary tumors (SPTs) constitute 8% to 12% of primary pancreatic tumors that develop at any age.1,2
There has been confusion in the SPT nomenclature regarding the macroscopic and microscopic characteristics of the tumor. The official term for this tumor is solid-pseudo papillary tumor3,4 although previously referred to as Frantz tumor, papillary cystic tumor, solid and papillary epithelial neoplasm, solid and/or cystic tumor, solid pseudopapillary epithelial neoplasm (SPEN) and papillary epithelial tumor. Since SPT is a relatively “new” phenomenon in pancreatic neoplasms, the true prevalence may have been partly concealed because the tumor was likely frequently misdiagnosed. Eight Chinese patients were studied by King, 5 in all cases, the preoperative clinical diagnosis was inaccurate, and in 4 of the patients, the initial postoperative diagnosis was “endocrine tumor.” V.K. Frantz identified this tumor as a novel entity that bears her name today in 1959 after compiling 4 examples that had previously been incorrectly identified as nonfunctioning islet cell tumors. 6
Researchers suggest that pluripotent embryonic stem cells with endocrine or exocrine potential may be the source of a tumor in teenage girls. The immunoprofile of SPT was associated with cells from the coelomic epithelium and the rete ovarii in a thorough immunohistochemistry analysis, indicating a close approach between the pancreas anlage and the left genital ridge during organogenesis. Nevertheless, no ovarian tumor that resembles SPT has been identified. This argument does not explain the lack of an ovarian tumor that resembles SPT, but it does support the prevalence of SPTs in teenage girls and their potential sensitivity to sex hormones.7,8 A case where fast tumor growth was seen during pregnancy could support this theory. 8
Method
This manuscript was written in accordance with SCARE requirements. 9
Ethical Approval and Informed Consent
The patient’s parents gave their written informed consent for this case report and its associated photos to be published. The Editor-in-Chief of this journal can evaluate a copy of the written consent upon request.
The office of institutional Research and Ethics Committee of St. Peter’s Specialized Hospital approved the case report with reference number of V987/16/5/2024.
Case Presentation
This is a 12-year-old child referred from the health center with the complaint of abdominal distension from 3 months ago (see Figure 1). Associated with this, she had nausea, vomiting of ingested matter, loss of appetite, and a non-quantified significant amount of weight loss. Since a month ago, the abdominal mass has increased in size rapidly, causing puritis, yellowish discoloration of the eye and skin, urine color change, and RUQ pain that radiates to the back. Otherwise, she has no history of contact with person with similar complaint and blood product transfusion. On physical examination she looks well, her vital signs are normal except her pain score which is +2, has muddy sclera and her abdomen distended and moves with respiration. There is a 15 cm by 20 cm firm, tender mass that covers the RUQ area and has a sharp edge. Negative for signs of fluid collection
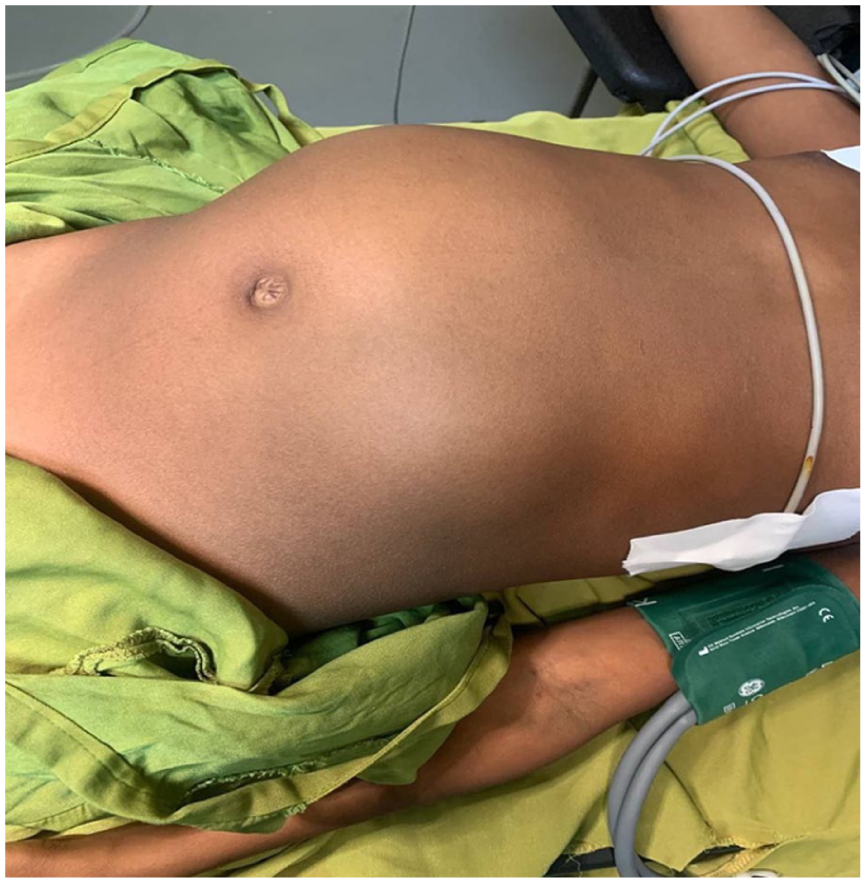
Twelve-year-old child with grossly distended abdomen (RUQ).
The laboratory and diagnostic results between March and June 2023 show complete blood count with differential were within the normal range, alongside progressive anemia with hemoglobin dropping as low as 7 g/dl and hematocrit down to 20%, though later improving to 11.5/32 following transfusion. Renal function tests, liver function tests and electrolytes are with in normal range, and pancreatic enzymes showed elevated amylase (279 U/L) but normal lipase (10.1 U/L). Viral markers (HBsAg, HCV, HIV) were non-reactive. Tumor markers (hCG B1=< 0.1 IU/ml, AFP = 1.7 ng/ml) were within normal ranges, and HVA was normal (4.28 mg/24 hours). ECG and chest X-ray were unremarkable, and COVID-19 rapid test was negative. And the Abdominal CT scan reveals there is a huge heterogeneous hypoattenuating lesion in the subhepatic space measuring 16.5 cm×14 cm that showed heterogeneous contrast enhancement in the arterial phase of the scan with mild increased enhancement of the center of the mass in the subsequent phases, but no internal calcifications seen. Mass appears to arise from the inferior surface of the liver. Mass displaced the pancreas anteriorly, and the head of the pancreas is not visualized. There is also mild dilation of the pancreatic duct. The gallbladder is compressed, and there is mild central biliary ductal dilatation. Otherwise, the liver has homogeneous attenuation and a smooth contour. The portal and hepatic veins are well enhanced with contrast and are patent, but the portal confluence is displaced and compressed. No peripancreatic or para-aortic lymphadenopathy seen. The spleen is normal in size and attenuation; no focal lesion is seen. With the conclusion of huge heterogeneously enhancing mass [? Arising from the head of the pancreas] suggesting solid pseudopapillary tumor of the pancreas (See Figure 2).
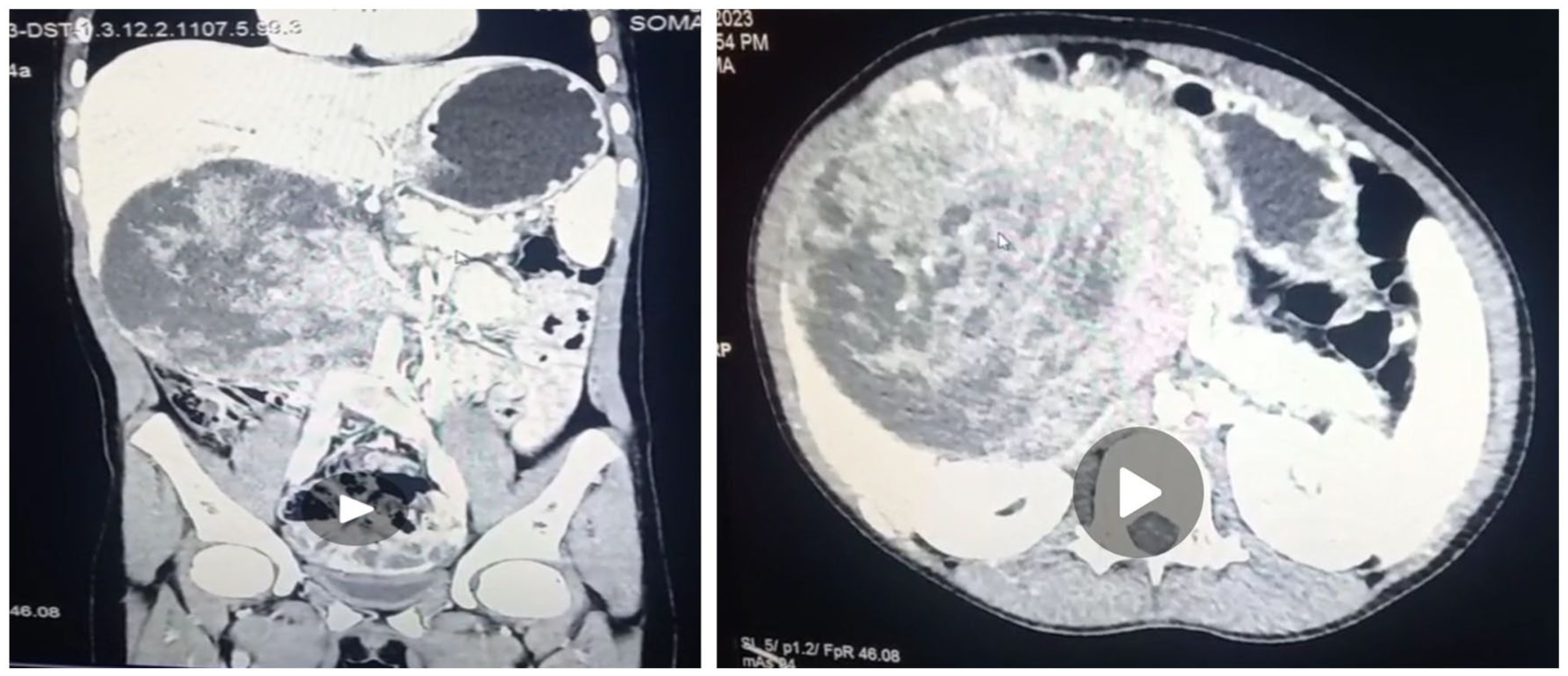
CT scan showed heterogenous hypoattenuating lesion in the subhepatic space.
Approximately 2 months later the patient underwent elective surgery revealing a huge cystic mixed with solid mass arising from head of the pancreas attached to the portal vein. Duodenum pushed anteriorly and medially rotated. Intra operatively she required 2 units of whole blood and fresh frozen plasma. Adrenaline also initiated and discontinued at OR. The duodenum was mobilized. The mass was mobilized internally, inferiorly, medially, and superiorly, separated from the portal vein, SMV, and SMA. GDA ligated. Complete resection (R0 resection) proximally from the pylorus, distal in location (jejunum resected out with duodenum) of the tumor with preservation of as much normal tissue as possible. Hepato-Jejunostomy (E-S), Gastro-Jejunostomy (S-E), and Pancreatic-Jejunostomy (E-S) were done. Drainage tube inside the GI and gastrojejunostomy stunt left. Cholecystectomy was done. The mass with the duodenum, jejunum, and CBD removed. See Figures 3 and 4.

Tumor approximately measuring 16.5 cm×14 cm located in the head of the pancreas.

Pancreatoduodenoctomy specimen with a macroscopic examination of the tumor.
Post operatively she was transfused with 1 unit of PRBC and transferred to ICU with stable vital sign for close follow up. After staying for 72 hours at ICU she was transferred to pediatric surgery ward with stable condition. The sample was sent for histopathology examination and reviled SPT (see Figure 5). For the last 9 month her post operate condition is stable no sign of recurrence and pancreatic insufficiency are present.
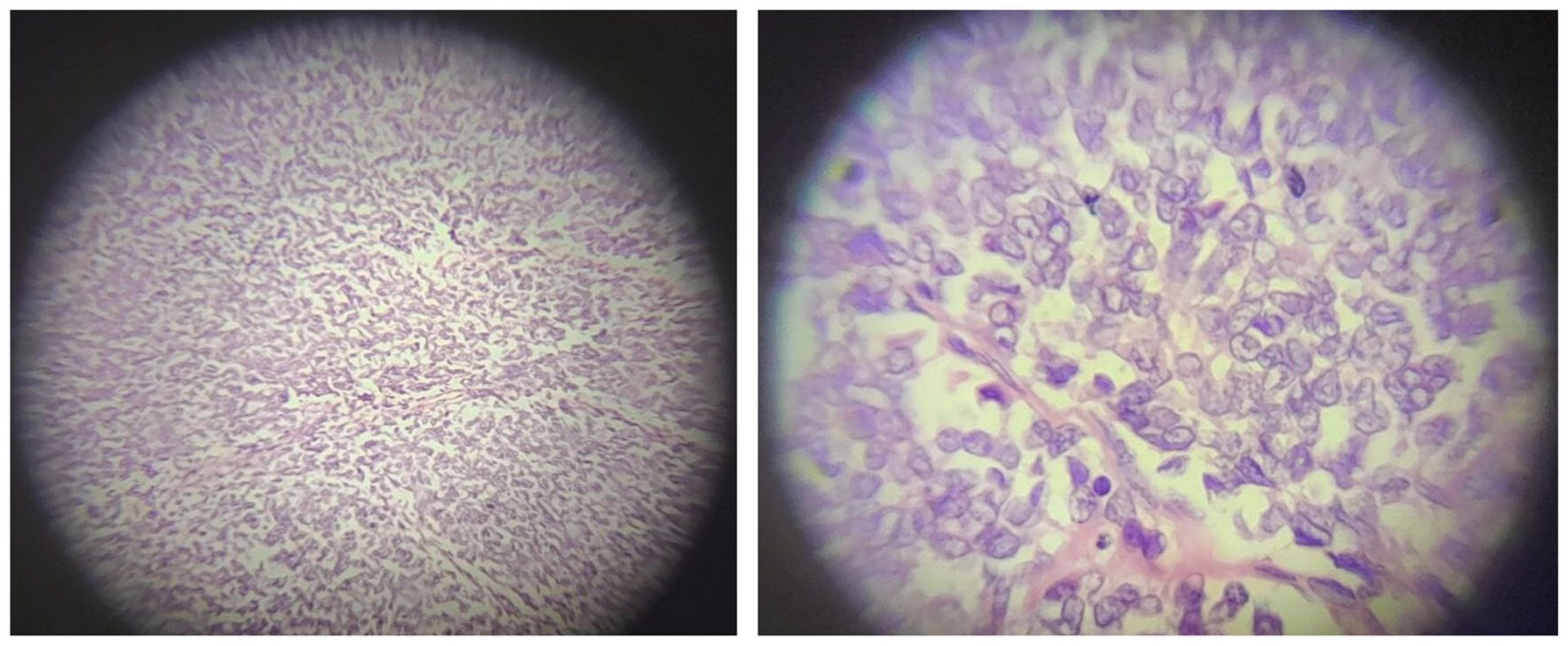
Histopathology of SPT (Post OP).
Discussion
In children, solid-pseudo papillary tumor of the pancreas (SPT) is an extremely unusual tumor. Its origin is still unknown. Ninety-one percent of cases are in young females and has a minimal chance for malignancy, particularly adolescent girls. There have only been a handful documented examples of male SPT patients to date. An uncommon primary pancreatic neoplasm, solid-pseudo papillary tumors (SPTs) constitute 8% to 12% of primary pancreatic tumors that develop at any age. 1 Compared to the head, the pancreas’s body and tail are more commonly affected (64%). Remarkably, tumors that were clearly growing outside of the pancreas were also seen 10 possibly as a result of ectopic pancreatic tissue. Pancreatic duct brush cytology with US-guided FNA may play a significant role in achieving a preoperative diagnosis because of their implications for surgical therapy. Frozen section biopsy is highly advised intra-operatively.
SPT is characterized by a benign clinical history and low malignant potential, despite potential histology evidence of malignancy. There are very few reports of childhood metastatic instances. 11 Multiple publications have highlighted that SPT is a surgically curable tumor, despite the extremely rare occurrence of metastases to the liver (7% of recorded patients), 5 lung, and skin. However, some cases may be locally aggressive and infiltrative (16% had major organ or blood artery invasion). There is research that males and older patients with malignancies are more prone to act aggressively.5,11 Additionally, there may be a higher proportion of Asian and Black patients. Rarely do metastatic tumors have aneuploid nuclear DNA, venous invasion, high nuclear grade, or noticeable necrobiotic nests, all of which indicate an increased risk of malignancy.7,12 -14
Risk factors for metastasis or recurrence include large tumor size (>8 cm), lymphovascular invasion, pancreatic parenchymal invasion, and a high Ki-67 index (>4%). 15 The Wnt/CTNNB1 pathway, leading to β-catenin accumulation, is a key molecular feature. 16
It is likely that patients with these characteristics should be actively monitored following surgery, even if good prognostic criteria are still lacking. It is surgical therapy that is preferred. Partial duodenopancreatectomy or distal pancreatectomy—ideally with the spleen preserved—are the common methods used to achieve complete removal of the tumor. Because of the tumor’s advantageous biological characteristics and the fact that the mass is usually surrounded by a dense fibrous capsule, tissue sparing surgery is both possible and justified. In certain situations, pylorus-preserving duodenopancreatectomy may be useful for tumor enucleation. Most evidence indicates that intensive local therapies or extensive lymphatic dissection are not advised. There is also widespread consensus that, notwithstanding the lack of standardization in metastasis treatment, surgical debulking (as opposed to alternative pancreatic malignancies) should be performed.
Establishing the histological diagnosis is crucial since complete surgical excision (R0 resection) of the tumor offers a good prognosis even in cases of metastases or local invasion. Currently, radiation therapy and chemotherapy do not have a well-defined role in primary treatment, but radiation has been used in the recurrent setting. 17 Systemic therapy, including mTOR inhibitors such as sirolimus and everolimus, which downregulate β-catenin, has shown promise in advanced cases. 18 Neoadjuvant and adjuvant chemotherapy have also been reported in pediatric SPT. 19
Long-term follow-up should monitor for pancreatic exocrine insufficiency, which can develop post-resection.
Conclusion
The body and tail of the pancreas are more frequently affected than the head in solid-pseudo papillary tumors of the pancreas (SPT), a very uncommon disease. Even in the event of metastases or local invasion, a full surgical excision of the tumor provides an excellent prognosis. molecular insights in to the Wnt/ β-catenin pathway and emerging systemic therapies offer a new avenue for managing advanced disease. Long-term follow-up should include for surveillances for pancreatic exocrine insufficiency.
Footnotes
Acknowledgements
We would like to thank all healthcare workers who are participated in the care of the patient.
Abbreviations
AFP: Alpha Feto Protein.
GDA: Gastroduodenal Artery.
ICU: Intensive Care Unit.
SMA: Superior Mesenteric Artery.
SMV: Superior Mesenteric Vein.
SPT: Solid-pseudo Papillary Tumor of the pancreas.
SPEN: Solid Pseudopapillary Epithelial Neoplasm.
Ethical Approval and Informed Consent
Written informed consent was obtained from the patient’s mother and father for publication of this case report and accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal on request.
Author Contributions
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authorship
Each author certifies that they fulfill the most recent ICMJE authorship requirements.
Data Availability Statement
The corresponding author will disclose the data upon reasonable request.*