
Abstract
A 3-year-old male patient with recurrent hospitalizations due to diarrhea, fever, vomiting, and seizures was discovered to have a GUCY2C gene mutation, inherited from his asymptomatic father. Despite antibiotics and supportive therapy, the patient continued to suffer from persistent secretory diarrhea, recurrent febrile seizures, and features of atypical Kawasaki disease. This case describes a rare presentation of early onset diarrhea and systemic inflammation associated with a gain-of-function mutation. The GUCY2C mutations are known to dysregulate intestinal fluid homeostasis, through excessive CFTR-mediated chloride secretion causing recurrent diarrhea. Additionally, this case suggests a potential link between gut barrier dysfunction and systemic inflammation, contributing to recurrent febrile episodes and KD-like manifestations. Standard antidiarrheal therapy was ineffective, highlighting potential roles for CFTR inhibitors and immune modulation. This case highlights the interplay between genetic mutations, gastrointestinal dysfunction, and systemic inflammation, underscoring the importance of a multidisciplinary approach in managing rare genetic enteropathies with immune involvement.
Introduction
The guanylate cyclase-2C gene, located on chromosome 12, encodes guanylate cyclase-C (GC-C), a transmembrane receptor activated by the endogenous hormones guanylin and uroguanylin, and by heat-stable Escherichia coli (E. coli) enterotoxin. It’s abbreviated as GUCY2C, other known abbreviations are GCC, GC-C, HSER, STAR, DIAR6, GUC2C, MECIL, and MUCIL. The gene sequence encodes a protein that activates the cystic fibrosis transmembrane conductance regulator (CFTR). Mutations in this gene have been linked to familial diarrhea (autosomal dominant pattern) and meconium ileus (autosomal recessive pattern). GC-C is important for regulating electrolyte, fluid secretion, and thus intestinal fluid homeostasis.1,2
In secretory diarrhea, the intestines secrete excessive amounts of water and electrolytes (such as sodium and chloride), leading to excessive fluid retention in the intestines and causing watery stools. Unlike other types of diarrhea, secretory diarrhea continues even during fasting because it is caused by active secretion rather than poor absorption. It can be triggered by infections, certain medications, toxins, or diseases affecting the gut lining. Patients usually have signs of abdominal cramping, urgency, and dehydration manifested by dry mucous membranes, decreased urine output, and electrolyte imbalances. Protracted diarrhea can reduce the quality of life of the patients and adversely affects nutritional status, causing physical fatigue. 3
The known genetic causes of congenital secretory diarrhea are mainly inherited in an autosomal recessive pattern. However, a single heterozygous mutation of the GUCY2C gene encoding GC-C was shown to be the cause of a dominantly inherited form of secretory diarrhea called familial diarrhea syndrome in 32 members of a Norwegian family. 4 The mutation described in this Norwegian study was a hyperactivating change found in the catalytic domain of GC-C. In particular, GC-C has heightened sensitivity to its endogenous ligands guanylin and uroguanylin, as well as the heat-stable enterotoxin of E. coli due to this mutation. This increased sensitivity leads to increased levels of cyclic guanosine monophosphate (cGMP) in epithelial cells, leading to increased fluid and ion secretion into the intestine and eventually diarrhea. Whereas inactivation of GUCY2C, as identified in 3 Bedouin families, caused meconium ileus resulting from impaired fluid and ion secretion into intestine that normally would be induced by the action of guanylin and uroguanylin on GC-C.5,6
The referred phenotype within the Norwegian family was similar to inflammatory bowel disease (IBD), with affected family members reporting 3 to 4 loose stools per day. Notably, 8 of the 32 affected individuals had severe dehydration since infancy. Moreover, 20% to 25% of these patients were diagnosed with small bowel obstruction and/or IBD. In contrast to this relatively mild presentation of diarrhea, the 4 patients with congenital secretory diarrhea described above had a more severe phenotype that always started in utero and was associated with early and severe IBD. All 4 of these individuals have distinct novel heterozygous de novo mutations in GUCY2C. These mutations are unique and localize to the catalytic domain of the receptor and to regions that allosterically regulate ligand-dependent activation of the guanylate cyclase domain. 4
The importance of this case lies in 2 things: the rarity of GUCY2C mutation and the recently increasing attention on the genetic features driving secretory diarrhea when such causes are typically thought to be caused by infectious or inflammatory mechanisms. This case report has been reported in line with the CARE (CAse REport) reporting guidelines. 7
Case Presentation
A 3-year-old Palestinian male patient presented to our emergency department following a generalized tonic-clonic seizure with frothy secretions and a rotated eye. The patient had a history of multiple hospitalizations.
The perinatal history was unremarkable, with no maternal history of hypertension, diabetes, urinary tract infections, or premature rupture of membranes. Birth history included that the patient was born at 36 weeks’ gestation via cesarean section due to maternal history of vertebral fracture. The birth weight was 2200 g, without a history of neonatal intensive care unit admission.
The family history was unremarkable, as none of his first-degree family members had ever complained of similar symptoms before, and there was no parental consanguinity. The patient did not undergo any surgery or take any long-term drugs, and he did not have any known drug or food allergies. Vaccination is up to date without complications. The feeding and nutritional history included formula feeding since birth, with poor appetite and feeding difficulties noted as he grew.
The patient’s medical history was significant for multiple hospital admissions, as his initial hospital admission occurred at 3 months of age, when he presented with a 1-month history of cough, cyanotic episodes, and poor feeding. Examination revealed retrognathia, a peaked nose, and mottled skin. He had bilateral wheezing and tachypnea, which raised the suspicion of bronchiolitis and possible sepsis. The patient’s symptoms were treated with nebulizers and antibiotics. After clinical improvement, the patient was discharged with salbutamol and normal saline nebulizers. However, 15 days after discharge, he started to have fever (39°C-40°C, measured axillary), which spiked multiple times daily and increased in frequency over time. It was associated with recurrent watery diarrhea occurring up to 14 times daily, which was non-bloody, non-greasy, and had no mucoid content; vomiting of gastric content without mucus or blood occurred after each feeding; hypoactivity; and poor feeding. Therefore, the patient was admitted as a case of gastroenteritis with sub-optimal level of consciousness. Three days after admission, he developed tonic-clonic-generalized seizures with cyanosis, which lasted for a few minutes. A full septic screen was performed due to prolonged fever, which revealed white blood cells (WBCs) (21 × 103/µL), platelets (333 × 103/µL), and hemoglobin (10 g/dL), and cerebrospinal fluid analysis (CSF) revealed bloody analysis, high protein (181 mg/100 mL), and high WBCs (negative after correction). The patient was treated for meningitis with ceftriaxone and acyclovir for 5 days. Urine analysis revealed a high white blood cell count, but urine culture after 3 days was negative. CSF culture revealed no growth, and blood culture revealed ESBL-Klebsiella, the patient was treated with amikacin and meropenem for 14 days. However, his fever persisted despite antibiotics, with negative control blood culture after the antibiotic regimen. A trial of stopping all medications was performed to determine if the fever was drug-induced, but it persisted after the trial.
The child had a history of papular skin rash on the face and abdomen, according to the mother, red cracked lips, and no history of joint pain or swelling. Further tests revealed a high platelet count (1 million/µL), high C-reactive protein (CRP) level, and high erythrocyte sedimentation rate (ESR).
After these findings and persistent fever despite antibiotics and antipyretics, prompt concerns for diagnosis of atypical Kawasaki disease (KD) as the patient complained of prolonged fever for more than 5 days, thrombocytosis, red cracked lips, papular rash on the abdomen, elevated CRP, and elevated ESR.
A cardiologist ordered an echocardiography, which revealed mild ectasia of the left main coronary artery (LMCA, 2.7 mm, Z-score 2.7) and the right coronary artery (RCA, 2.8 mm, Z-score 3.5). Additionally, a small patent foramen ovale (PFO) was identified. No intracardiac masses, vegetations, or pericardial effusions were observed. Color Doppler study revealed trace tricuspid and mitral valve insufficiencies with normal pulmonary arterial systolic pressure and no evidence of valvular stenosis.
Intravenous immunoglobulin (IVIG) was administrated in 2 divided infusions over 2 days, 10 g per day, along with high dose aspirin (100 mg, every 6 hours) to treat and prevent complications. Following this regimen of IVIG and aspirin, the patient became afebrile.
Six months later, follow-up echocardiography showed no further signs of coronary artery ectasia (LMCA: 2.1 mm, RCA: 2 mm), indicating regression of the previous dilation. The cardiac chambers, valves, and ventricular septum remained structurally normal, and the outflow tracts were patent. Although the PFO persisted, it remained small. No new pathological findings, such as pericardial effusion or intracardiac masses, were observed. The diagnosis was incomplete (atypical) KD, which occurs more commonly in infants younger than 6 months of age and does not meet the full diagnostic criteria of KD, as discussed later.
Genetic testing through whole exome sequencing (WES) identified that the patient was heterozygous for the variant c.3049G>A, p.E1017K in exon 27 of GUCY2C gene, which is suspected to contribute to early-onset recurrent diarrhea and systemic inflammatory disease. The WES test conducted on the patient’s parents revealed that the father was heterozygous for the variant (c.3049G>A, p.E1017K) in exon 27 of GUCY2C gene, while the mother was normal. Figure 1 presents the family pedigree, illustrating the inheritance of GUCY2C gene mutation.

Genetic pedigree illustrating the inheritance of GUCY2C gene mutation.
At 5 months old, he was admitted because of recurrent diarrhea, occurred 8 times daily, which was non-bloody, and non-mucoid. This was associated with vomiting and fever. The patient was treated with oral rehydration therapy and antipyretics
At 2 years old, the patient exhibited a persistent high-grade fever (up to 40°C, axillary) for 15 days, which improved with antipyretics. This was complicated by a generalized tonic-clonic seizure lasting 4 minutes, characterized by up-rolling eyes, with no history of vomiting, diarrhea, loss of consciousness. Examination revealed irritability, pharyngitis, and prolonged fever, along with papular lesions on the left wrist in a dermatomal pattern, suggesting a herpes virus infection; thus, IV acyclovir was administrated. Laboratory tests revealed normal CSF analysis, CRP (15 g/L), ESR (25 mm/hour), lactate dehydrogenase (250 U/L), negative cultures, and a negative viral panel. Given the history of atypical KD, an echocardiography was performed with no evidence of coronary ectasia and good cardiac function. Additionally, tests for brucella, antinuclear antibodies, and rheumatoid factor were negative, and an abdominal ultrasound showed no signs of hepatosplenomegaly, or abscess.
At 3 years old, the patient presented to our emergency department following a generalized tonic-clonic seizure characterized by frothy secretions and rotated eye. The seizure lasted a few minutes and resolved spontaneously. It was accompanied by a fever of 40°C (measured rectally), cyanosis around the mouth, and loss of consciousness.
Upon examination in the emergency department after the seizure, the patient was in a postictal state, appearing drowsy but responsive, with mild dehydration but no signs of respiratory distress. A neurological examination revealed that the patient was alert after the seizure, with normal muscle tone, power, and reflexes. Examination of the other systems was normal. His vital signs: Temperature (39.2°C, axillary), heart rate (145 bpm), respiratory rate (30 breaths/min), and oxygen saturation (96%, on room air).
The patient was treated with intravenous (IV) fluids for dehydration, ceftriaxone (500 mg, every 12 hours, IV), paracetamol (150 mg, IV) for fever control.
Figure 2 presents a clinical timeline that summarizes the case presentation and clinical events in our patient.

Clinical timeline figure.
Discussion
This case highlights a unique and rare instance of a gain-of-function mutation in the GUCY2C gene, which contributes to early onset diarrhea and systemic inflammatory disease. The GUCY2C encodes the GC-C receptor, a critical regulator of intestinal fluid homeostasis. It is activated by endogenous ligands guanylin and uroguanylin, as well as bacterial heat-stable enterotoxins, leading to the activation of CFTR channels and subsequent chloride and bicarbonate secretion into the intestinal lumen. While loss-of-function mutations in GUCY2C are known to cause intestinal pseudo-obstruction and constipation. Gain-of-function mutations have been increasingly associated with persistent secretory diarrhea syndromes. 4
Secretory diarrhea results from excessive intestinal fluid secretion, independent of oral intake, and is commonly attributed to infectious, inflammatory, or toxin-mediated causes. However, genetic disorders leading to monogenic diarrheal syndromes have gained recognition, particularly those involving GUCY2C mutations. 8 The c.3049G>A, p.E1017K variant of GUCY2C gene identified in our patient has not been previously reported in the medical literature or databases as a direct cause of diarrhea, making it a novel finding. However, other mutations and variants in the GUCY2C gene have been associated with increasing the intracellular cGMP signaling. This increase likely led to hyperactivation of CFTR, which when stimulated by cGMP, result in increased chloride and water in the intestine, explaining its potential to cause secretory diarrhea.4,9 This variant may be the cause of diarrhea through its effect in enhancing the cGMP signals, leading to persistent activation of CFTR-mediated chloride secretion and severe diarrhea. Unlike transient infectious diarrhea, genetic diarrhea syndromes are chronic, refractory to conventional treatments, and require a targeted therapeutic approach. 10
Recent studies indicate that GC-C signaling is not confined to the gastrointestinal tract, but also plays a role in modulating inflammatory pathways and immune homeostasis. 11 Dysregulated GC-C activity may contribute to excessive pro-inflammatory cytokine release, immune dysregulation, and recurrent febrile episodes, as observed in this patient. Additionally, the association of infections, such as Klebsiella pneumoniae with prolonged inflammatory responses suggests an underlying immunological vulnerability that warrants further investigation. 12
The interplay between chronic intestinal inflammation and systemic immune activation remains an area of active research, with emerging evidence suggesting that gut barrier dysfunction may trigger systemic inflammatory responses through microbial translocation and immune modulation. 13 Kawasaki-like symptoms in our patient is more likely accounted for by the convergence of genetic enteropathy, barrier dysregulation, and infection-related triggers. Zonulin is a structural and functional related proteins released from epithelial cells of gastrointestinal tract, which reversibly regulate intestinal permeability by modulating intercellular tight junctions. Several diseases including autoimmune and hyperinflammatory diseases such as KD, celiac disease, and IBD have been showed increased circulatory zonulin levels. In Multisystem Inflammatory Syndrome in Children (MIS-C) and hyperinflammatory diseases, zonulin-mediated intestinal breakdown promotes increased permeability, permitting microbial and antigenic translocation through into circulation, driving systemic cytokine storms and endothelial injury. GUCY2C gain-of-function in our case leads to chronic enteropathy and impaired mucosal defense, with superimposed infections such as Klebsiella leading to additional antigenic challenge that exacerbates systemic immune activation. This convergence of genetic predisposition, intestinal barrier breach, and infectious exposure could explain the vasculitic and hyperinflammatory phenotypes mimicking Kawasaki-like presentation. Putting the patient’s case into the larger context of MIS-C and other post-infectious inflammatory syndromes underscores the intestine’s pivotal position as both barrier and immunological driver of systemic vasculitis-like disease.14,15
Given the genetic basis of the patient’s condition, standard anti-diarrheal agents, such as loperamide, may be ineffective, as they do not target the underlying CFTR-mediated secretory mechanism. 16 Current therapeutic strategies under investigation include CFTR inhibitors, such as crofelemer, which could reduce chloride secretion and stool volume. 17 A recent case report by William Scott et al. described a patient with a novel GUCY2C variant who was treated with crofelemer with partial response, providing one of the first real-world clinical demonstrations of its potential utility in congenital secretory diarrhea, it highlights both the therapeutic promise and the need for further optimization of CFTR inhibition. 18 Other strategies include targeted GC-C modulators, which may help regulate excessive intestinal secretion; electrolyte and fluid management to prevent dehydration and electrolyte disturbances; and dietary modifications, including the use of an elemental or low-FODMAP (fermentable oligosaccharides, disaccharides, monosaccharides, and polyols) diet, to minimize gut irritation. 17
A particularly intriguing aspect of this case is the co-occurrence of systemic inflammatory episodes and atypical KD-like symptoms. One exciting hypothesis is that chronic enteropathy with GUCY2C gain of function mutations might trigger KD-like inflammation by breaching the intestinal barrier. In particular, secretory diarrhea may contribute to microbial translocation, resulting in chronic immune activation by luminal antigens. Such chronic immune activation may then lead to the characteristic KD-like inflammation, with endothelial damage and hypercytokinemia, as a pathological mechanism. Furthermore, aberrant GC-C signaling is known to influence immune modulation. This suggests that barrier and signaling defects, giving rise to a hyper-inflammatory state. This intersection in mechanisms may explain the diarrhea and Kawasaki-like features observed in our patient. 15
The diagnosis of KD is determined by the presence of a fever for more than 5 days, excluding of other diseases causing similar findings, and accompanied by at least 4 of the following 5 symptoms: (1) bilateral nonsuppurative conjunctivitis; (2) one or more changes of the mucous membranes of the upper respiratory tract, such as pharyngeal injection, dry fissured lips, injected lips, and a “strawberry” tongue; (3) one or more changes of the extremities, including peripheral erythema, peripheral edema, periungual desquamation, and generalized desquamation; (4) a polymorphous rash, primarily truncal; (5) posterior cervical lymphadenopathy greater than 1.5 cm in diameter. 19
Atypical KD, which is more common in infants younger than 6 months of age, present with clinical features suggestive of KD but does not meet the full diagnostic criteria. Consider atypical KD in: (1) a child with fever for at least 5 days combined with 2 or 3 of the principal clinical criteria, especially, if coronary artery aneurysms are presents; (2) an infant with one or more of the following features: fever ≥ 7 days with/without irritability without other explanation, or prolonged fever and unexplained aseptic meningitis; (3) a child or infant with prolonged fever with shock or cervical adenitis not responsive to oral antibiotics.20,21 Our patient exhibited prolonged fever of more than 5 days, red cracked lips, and polymorphic rash, yet did not meet the full KD diagnostic criteria. Additionally, thrombocytosis, elevated CRP, and elevated ESR support the diagnosis of atypical KD. Since many of the manifestations of KD are found in other illnesses, such as infections, inflammatory conditions, and drug reaction, these must be considered and excluded before establishing a KD diagnosis. The initial blood culture revealed Klebsiella, which was considered a cause of fever, but fever persisted despite a sensitive antibiotic regimen and a negative control blood culture after antibiotic treatment. A drug reaction was considered, but the fever persisted after the discontinuation of all medications to rule out a drug reaction. The diagnosis of KD was more likely to be the cause of prolonged fever and inflammatory conditions in the patient. Additionally, the finding of coronary artery ectasia clinched the diagnosis of atypical KD despite incomplete clinical criteria.20,21
KD rarely coincides with infectious processes, and several reports have described its occurrence alongside bacterial or viral infections. The coexistence of infection and KD presents a diagnostic challenge, as fever and inflammatory changes may be attributed to the infectious process.22,23 The atypical KD in our patient was triggered by or coincided with infection due to the multiple history of infections.
Interestingly, the father of the patient was heterozygous for the same mutation but remained asymptomatic, suggesting variable expressivity and incomplete penetrance, which are features observed in other autosomal dominant gastrointestinal disorders. This highlights the need for genetic evaluation in cases of unexplained persistent diarrhea, particularly in pediatric patients with an unclear diagnosis.
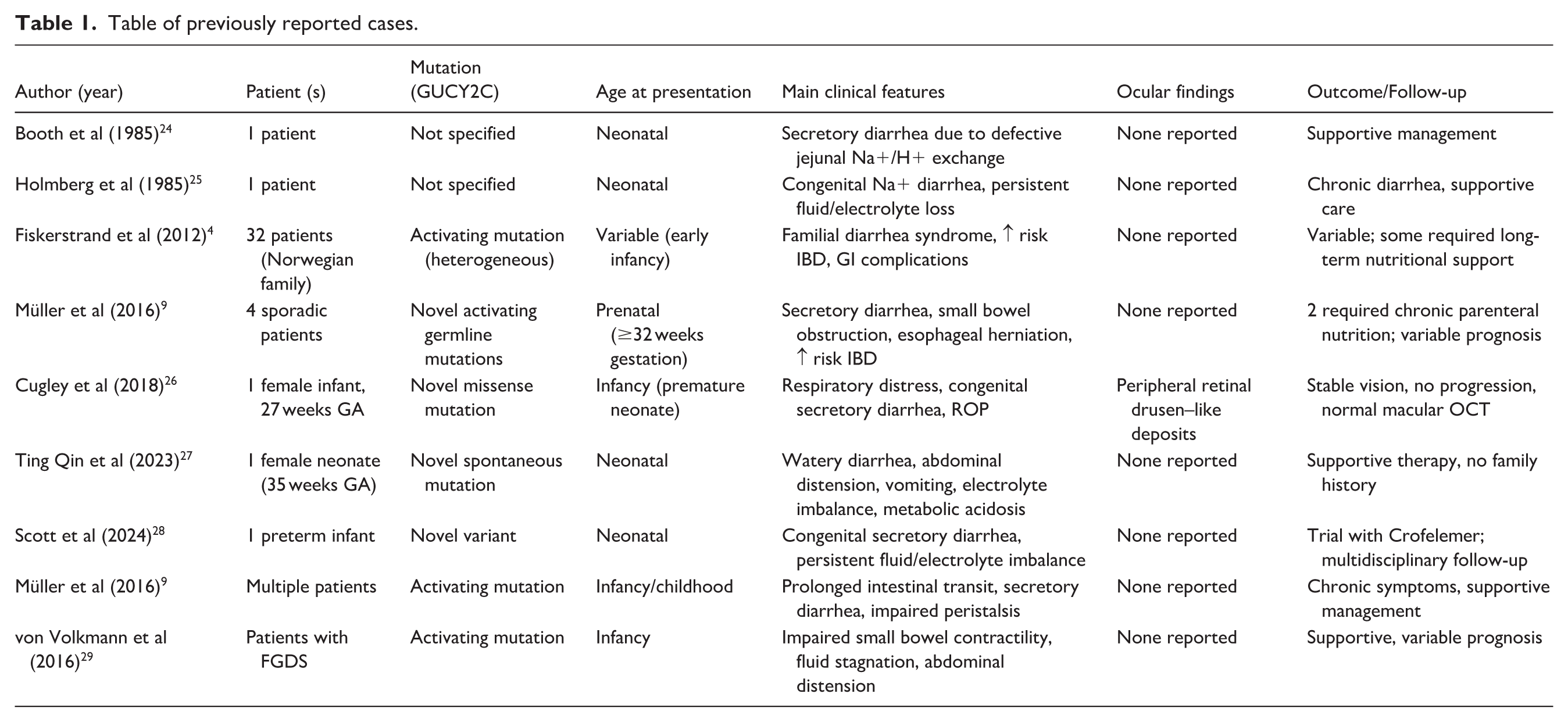
Table 1 shows previously reported cases of secretory diarrhea, some of which were caused by GUCY2C mutations at different ages of presentation.
Table of previously reported cases.
The recurrent inflammatory episodes in this patient underscored the need for close monitoring, serial inflammatory markers, and immunological evaluation. Future studies are needed to explore the potential of targeted therapies that modulate GC-C signaling in patients with genetic diarrhea syndromes. To our knowledge, this is the first reported case of a GUCY2C gene mutation in Palestine.
Conclusion
This case highlights the complex cross-section between genetic mutations, gastrointestinal dysfunction, and systemic inflammatory responses. GUCY2C gain-of-function with the heterozygous c.3049G>A, p.E1017K mutation led to persistent secretory diarrhea, exacerbating dehydration, electrolyte imbalances, and recurrent hospitalizations.
Early genetic testing is critical in detecting mutations and guiding care, emphasizing its importance in recognizing unexplained pediatric diarrhea.
Furthermore, the atypical presentation of KD made the diagnosis of GUCY2C mutation difficult, early IVIG therapy was given to prevent cardiac problems, which were eventually resolved on follow-up. Recurrent febrile seizures exacerbate the situation, emphasizing the need for multimodal therapy to address neurological concerns and systemic inflammation.
Long-term surveillance is required to examine growth, heart status, and potential immunological dysregulation. This case emphasizes the necessity of putting genetic testing into standard diagnostics for chronic pediatric gastrointestinal illnesses, as well as early detection and prompt care of inflammatory syndromes, such as atypical KD.
Footnotes
Acknowledgements
The completion of this case could not have been possible without the participation and assistance of many people whose names may not be enumerated like our patient’s parents, their contributions are sincerely appreciated and gratefully. Additionally, we would like to thank Dr. Husien sarahneh for his valuable assistance in reviewing the case report for language clarity and polishing. His support is sincerely appreciated. We would like to express our sincere gratitude to all the healthcare professionals who contributed to the successful diagnosis and management of this case.
ORCID iDs
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent for Publication
Written informed consent was obtained from a legally authorized representative (parents) for anonymized patient information to be published in this article.
Author Contributions
Writing the original draft: Mohamad A. Banat, Mahmoud Jawabreh, Qusai Mahmoud Belbaisi, Bara Atawneh, Hamza M. A. Ghaith.
Reviewing & editing the manuscript: Mohamad A. Banat, Ramzi Mujahed, Mahmoud Jawabreh, Qusai Mahmoud Belbaisi, Bara Atawneh, Hamza M. A. Ghaith.
Contributed to conception or design: Mohamad A. Banat, Ramzi Mujahed.
Contributed to acquisition, analysis, or interpretation: Mohamad A. Banat, Mahmoud Jawabreh, Qusai Mahmoud Belbaisi, Bara Atawneh, Hamza M. A. Ghaith.
Gave final approval: Mohamad A. Banat, Ramzi Mujahed, Mahmoud Jawabreh, Qusai Mahmoud Belbaisi, Bara Atawneh, Hamza M. A. Ghaith.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.