
Abstract
Paratesticular rhabdomyosarcoma (RMS) is a rare tumor. Imaging helps diagnose the tumor process, while histopathological examination with immunohistochemistry confirms the diagnosis. Treatment should be multimodal, involving surgery, chemotherapy, and radiotherapy. In light of this observation and a review of the literature, discuss a case of paratesticular rhabdomyosarcoma with invasion of the ipsilateral testicle.
Introduction
Paratesticular rhabdomyosarcoma is a rare and aggressive tumor, accounting for approximately 7% of all childhood rhabdomyosarcomas. 1 Among urogenital lesions, the paratesticular location is the most common. It includes several subtypes, with the embryonal variety being the most prevalent. The prognosis is generally poor, and the differential diagnosis often includes scrotal emergencies such as testicular torsion and post-traumatic scrotal hematoma. Due to its rarity and clinical resemblance to more common scrotal emergencies, paratesticular rhabdomyosarcoma remains a challenging diagnosis.
Diagnosis is typically suspected through ultrasound and confirmed by histopathological examination of the surgical specimen following orchiectomy. This malignancy has a poor prognosis, and its treatment generally involves a combination of surgical resection, chemotherapy, and, in some cases, radiotherapy. Early diagnosis and prompt treatment are essential to improve patient outcomes. 2
By consolidating key imaging and histopathological findings, we aim to enhance the understanding of this aggressive tumor and highlight the importance of early detection and multimodal management.
To the best of our knowledge, this is the first documented clinical case of paratesticular rhabdomyosarcoma with almost total invasion of the testis.
Case Report
A 5-year-old child presented with a large left scrotal mass that had developed over less than 3 weeks; there was no significant medical history. The reason for the pediatric emergency appointment was left testicular pain that had started at least 5 months ago. Upon clinical examination, the patient appeared in overall good health, afebrile, with an enlarged left testicle showing regular contours, firm consistency, no palpable tenderness, and no signs of local inflammation. There was no gynecomastia or lymphadenopathy, and the rest of the clinical examination was unremarkable. Transillumination test results were negative. Testicular ultrasonography revealed a heterogeneous oval hypoechoic solid mass measuring 29 mm × 28 mm, with vascularization evident on color Doppler imaging. It showed well-defined borders and a fuzzy margin with the ipsilateral testicle, causing compression of the latter (Figure 1). Absence of secondary localization was detected in the thoraco-abdominal-pelvic or pelvic computed tomography, including no retroperitoneal ormediastinal lymphadenopathy or hepatic metastases. The left scrotum showed a mixed cystic and solid mass at the pelvic level, increasing in size. It had an indistinct boundary, and the ipsilateral testicle associated with it measured 39 mm × 28 mm (Figure 2).
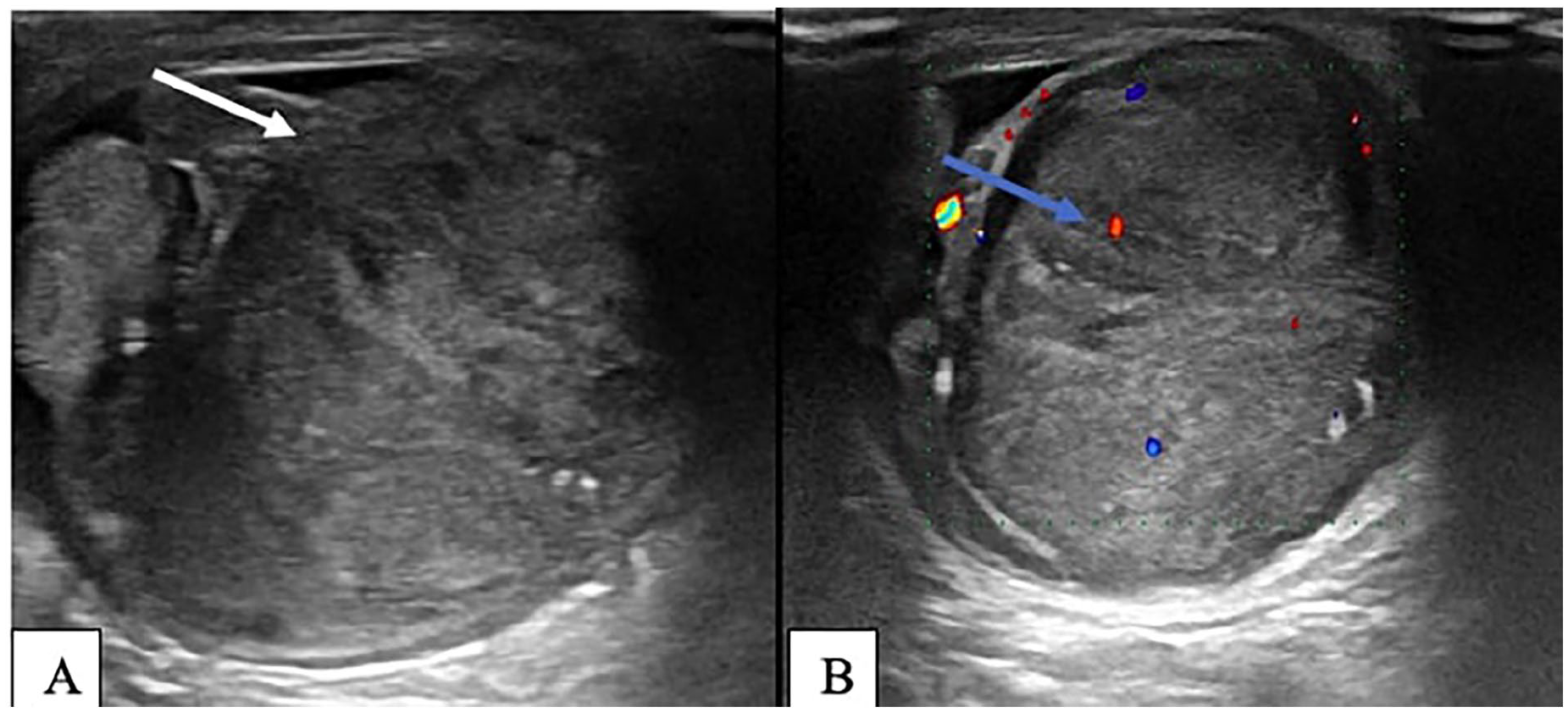
Heterogeneous, oval, hypo-echoic solid mass (A) with regular contours, hyper-vascularized on color Doppler blue arrow (B).

Left purse increased in size seat of a mixed cystic and tissue process with fuzzy border with the ipsilateral testicle (A) with peripheral enhancement after PDC injection (B).
This case was discussed during a multidisciplinary tumor board meeting, which included specialists from radiology, pediatric oncology, pathology, and surgery. The team collaboratively decided on the surgical approach with high ligation of the spermatic cord followed by inguinal orchiectomy. Given the diagnosis of embryonal paratesticular rhabdomyosarcoma, the IVA chemotherapy protocol was initiated. Regular follow-up imaging and clinical monitoring were also planned to ensure early detection of any recurrence or metastasis.
Following high initial ligation of the spermatic cord, an inguinal orchiectomy was performed. The tumor occupied almost the entire testicular parenchyma, with a small remnant of normal tissue. It presented with 2 distinct zones: a cystic zone that breached the tunica vaginalis near the solid zone, with leakage of citrine yellow serous fluid. The solid zone was firm and yellow in color.
The macroscopic appearance of the tumor, which takes up almost the entire testicular parenchyma, includes a solid area of firm consistency and yellowish coloration, as well as a cystic area characterized by the discharge of a clear citrine yellow fluid with Proliferation Index: 40% in the most active areas, indicating high proliferative activity.
A histological analysis of the orchidectomy tissue reveals a mesenchymal growth composed primarily of rounded and fusiform cells with elongated nuclei, small nucleoli, and reduced eosinophilic cytoplasm (Figure 3A). The immunohistochemical study shows positive staining for Myo D1 and myogenin positive in 30% of cells (Figure 3B and C) and desmin positive focal (Figure 3D). These cells demonstrate rhabdomyoblastic differentiation, which is absent in the remaining testicular parenchyma. In the paratesticular area, there is a 7 cm long axis of fusiform cells indicating embryonic rhabdomyosarcoma.

Pathology image of paratesticular rhabdomyosarcoma: Mesenchymal proliferation with cells sometimes spindle-shaped, sometimes round, showing marked cytonuclear atypia; the stroma is edematous and myxoid (A), Positive staining for myo D1(immunohistochemistry) (B), myogenin positive in 30% of cells. (immunohistochemistry) (C), Desmin Positive Focal (D).
Following the surgical resection, the patient was started on adjuvant polychemotherapy with the IVA protocol (Ifosfamide, Vincristine, Actinomycin D). The chemotherapy was administered in cycles, with Ifosfamide given intravenously every 3 weeks, Vincristine weekly, and Actinomycin D on day 1 of each cycle, for a total of 6 cycles. The patient tolerated the treatment well with mild neutropenia, nausea, vomiting, alopecia and fatigue, all of which were managed appropriately. Postoperatively, the patient recovered well from the inguinal orchiectomy, with no complications such as bleeding or infection. The incision site healed without any issues, and the patient was discharged 5 days after surgery. Regular follow-up included clinical evaluations, ultrasound imaging, and chest X-ray to monitor for recurrence or metastasis. The chemotherapy regimen was well-tolerated, and the patient will continue follow-up for 2 years, with evaluations every 4 months during the first year and every 6 months thereafter.
Discussion
Paratesticular rhabdomyosarcoma (PRMS) is a rare tumor with 2 primary peaks of incidence: the first between 2 and 5 years of age and the second during adolescence. 3 It is believed to originate from mesenchymal elements of the tunica vaginalis of the testis, epididymis, or spermatic cord. While compression of the testicle by the mass is common, invasion of the ipsilateral testicle is exceptionally rare, with no cases documented in the literature.5,6
Paratesticular rhabdomyosarcoma is characterized by rapid progression, occurring most commonly in children and young adults, with incidence peaks at 4 years and again at 16 years. 4 No racial predisposition has been identified, and local extension occurs early, with dissemination via lymphatic and hematogenous routes. 5 Recent reviews also highlight the importance of early detection in improving prognosis, as metastasis to regional and distant sites (eg, lungs, liver, and retroperitoneal lymph nodes) is common.6,7
Clinically, Paratesticular rhabdomyosarcoma presents similarly to other intrascrotal tumors, which makes it difficult to differentiate based on physical examination alone. It is typically a firm, non-painful mass with rapid progression, affecting either side of the scrotum. 5 Tumor markers specific to PRMS are lacking, and diagnosis relies entirely on histological examination of the orchidectomy specimen. Histologically, PRMS can present in 3 main subtypes: embryonal (the most common, comprising 95% of cases), alveolar, and pleomorphic, each with distinct age predilections. 6 The embryonal type is predominantly observed in children, while the alveolar and pleomorphic types are more common in adolescents and adults. 8
Recent studies emphasize the role of immunohistochemical staining for confirming the diagnosis of PRMS. Positive staining for desmin and myogenin, markers of muscle differentiation, is highly suggestive of the diagnosis. 9 Our patient’s case is unique in that it involves the ipsilateral testicle, which, to our knowledge, has not been previously reported in the literature. This rare feature highlights the complexity of diagnosing and treating PRMS in unusual presentations.
Testicular ultrasound remains the first-line imaging modality, showing a heterogeneous mass with inguinoscrotal extension in 80% of cases. 7 Doppler ultrasound can also aid in identifying hypervascular regions, which may be indicative of malignancy. The challenge of distinguishing PRMS from other scrotal tumors emphasizes the need for high-quality imaging techniques, including MRI, which can provide additional insights into the extent of the mass and its relation to surrounding structures. 10 In our case, the mass’s boundary with the testicle was unclear, necessitating further investigation, in our case, boundaries with the ipsilateral testicle were hazy, making it uncertain whether the tumor originated in the testicle and extended to the bursa level or if it originated extratesticularly and invaded the ipsilateral testicle.
CT imaging of the thorax, abdomen, and pelvis is critical to assess for lymph node involvement and distant metastasis, particularly in high-risk cases. The retroperitoneal nodes are often the first to be affected, and lymphatic spread is documented in 26% to 43% of patients with PRMS. 6 Pulmonary and hepatic metastases are also common, and the detection of these lesions is essential for staging and treatment planning. MRI with surface coils, showing distinct T2 signal intensities, can help differentiate PRMS from normal testicular tissue. 8
The treatment of PRMS generally involves radical inguinal orchidectomy with cord ligation, which serves both therapeutic and diagnostic purposes. Post-surgical treatment depends on the disease stage and may include chemotherapy, radiation therapy, and retroperitoneal lymph node dissection.9,10 Chemotherapy regimens, including vincristine, actinomycin D, and cyclophosphamide, have been shown to improve survival rates and reduce relapse in high-risk variants. 11 Recent studies support the use of combined therapies for advanced cases, as this multimodal approach has yielded better survival outcomes.9,12
In our case, inguinal orchidectomy was performed, and staging showed no evidence of metastasis, allowing us to forgo chemotherapy and adopt a strategy of observation. However, given the risk of recurrence, ongoing monitoring with imaging and clinical evaluation is essential.
Differential diagnoses for PRMS include liposarcoma, leiomyosarcoma, and fibrosarcoma, which are more commonly seen in adults. These tumors present similarly on imaging, making histology the definitive tool for diagnosis. 8 The rarity of testicular invasion by PRMS further complicates the differential diagnosis and underscores the importance of a thorough histological assessment.
Conclusion
Testicular invasion in PRMS represents an exceptionally rare presentation of an already uncommon tumor. This highlights the importance of early interven-tion for both diagnostic and therapeutic purposes. Multimodal treatment approaches, including surgery, chemotherapy, and radiation therapy, have shown promising survival outcomes, especially when managed through a multidisciplinary approach. Accurate staging, early diagnosis using imaging and clinical examination, and a structured treatment protocol are critical for improving prognosis. Long-term follow-up, including regular imaging and clinical evaluations, remains essential for ensuring optimal patient management and surveillance.
Footnotes
Acknowledgements
I would like to express my gratitude to my professors and all the colleagues who participated in the completion of this work.
Ethical Considerations
Our institution does not require ethical approval for reporting individual cases or case series.
Consent to Participate
Written informed consent was obtained from the parents of patients for the publication of this case report.
Author Contributions
All authors equally contributed to the collection of information, data analysis, and manuscript writing. All authors have read and approved the final version of the manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.