
Abstract
Subacute sclerosing panencephalitis (SSPE) is a rare, fatal encephalitis caused by chronic measles virus infection. Its recent resurgence, particularly after measles epidemics, underscores the need for vigilance in children. This study describes the clinical, paraclinical, therapeutic features, and prognosis of SSPE in 15 patients. Initial symptoms included gait disturbances, psychomotor regression, and behavioral changes. Neurological examination revealed cognitive decline, visual impairment, hypotonia, pyramidal signs, ataxia, and movement disorders. Electroencephalogram showed periodic delta wave complexes, while brain imaging identified subcortical and periventricular white matter (WM) lesions, brainstem involvement, and progressive cortical-subcortical atrophy on follow-up. All patients tested positive for anti-measles immunoglobulins G in cerebrospinal fluid and blood. Despite treatment, rapid clinical deterioration led to akinetic mutism and neurovegetative symptoms. This study alerts clinicians and radiologists to consider SSPE in children presenting with subacute cognitive decline, myoclonus and epilepsy with periodic complexes, associated with specific WM changes, particularly in the post-epidemic period.
Introduction
Subacute sclerosing panencephalitis (SSPE) is a rare and fatal, subacute encephalitis caused by latent measles virus that occurs years after primary infection. 1 It causes widespread demyelination of the central nervous system, commonly seen in children and young adults. 2 The clinical, electroencephalographic and radiological features are not particularly specific and can sometimes lead to a misdiagnosis. Symptom onset is often insidious with behavioral and cognitive deterioration, myoclonic seizures, and progressive motor impairment. It generally affects children who contracted the measles virus at a young age, particularly before vaccination. 3 A measles epidemic has broken out in the Maghreb region in 2019, with 4669 cases in 2019. 4 Consequently, over the past 4 years, we have seen an upsurge in cases of SSPE in our department. The reemergence of this disease in recent years calls for particular vigilance among children, especially in developing countries where measles is still endemic. There is limited data on SSPE in North Africa particularly in Tunisia.
Objective
To analyze epidemiological, clinical and paraclinical features of SSPE in Tunisian children who contracted measles during the 2019 outbreak.
Methods
We conducted a retrospective and descriptive study over a period of 4 years between August 2020 and April 2024 including children meeting Dysken’s SSPE criteria. 5 A case of SSPE is defined as an illness with a compatible clinical course plus 1 supporting laboratory evidence such as detection of measles antibody in the cerebrospinal fluid (CSF), a characteristic pattern on electroencephalography (periodic complexes of bilaterally synchronous, and symmetrical 1-3 Hz waves or sharp and waves) or typical histologic findings in brain biopsy material or tissue obtained on postmortem examination or specialized molecular diagnostic test identifying wild-type measles virus mutated genome.
Disease onset was considered rapidly progressive if it developed in less than a month, and subacute otherwise. Acute fulminant SSPE was defined by rapid progression of the disease, leading either to death or an akinetic-mute state within 6 months of symptoms onset. On admission, patients were classified into stages according to Jabbour et al
6
classification.
All patients underwent brain magnetic resonance imaging (MRI), which was reviewed by an experienced neuroradiologist. Scalp electroencephalography was recorded using the international 10 to 20 system. Measles antibodies in blood and CSF were detected by Enzyme-linked immunosorbent assay (ELISA) in all patients.
This study is a retrospective analysis of subacute sclerosing panencephalitis cases. Ethical approval was not required as the research involved the analysis of existing data without direct interaction with human subjects. Therefore, informed consent was also not necessary. The study was conducted in accordance with international ethical guidelines, ensuring confidentiality and anonymity of the data used.
Data relating to demographic, clinical, biological and electrophysiological features, neuroimaging findings, therapeutic management, and outcome, were collected.
Results
Fifteen unrelated patients (12 boys and 3 girls) were included. Their home countries were Tunisia (10/15), Algeria (4/15) and Libya (1/15). None of the patients had received vaccination against measles prior to infection. The mean age at symptom onset was 45 months [21-73], while the mean age at the time of measles virus infection was 6 months [1-12]. The interval between measles infection and symptom onset ranged from 13 to 61 months. The disease was rapidly progressive in 4 cases and subacute in 11 patients. Six patients had acute fulminant SSPE. In 12 cases, a banal infection preceded the symptom onset by an average of 17 days. The diagnosis was established at stage II in 8/15 patients, stage III in 6/15 patients, and stage IV in 1/15 patient. The average time to diagnosis was 74 days [7 days − 8 months].
Clinical Findings
Gait disturbances, psychomotor regression and behavioral changes were the most common inaugural symptoms in our patients. The main clinical findings on admission are represented in Table 1. Movement disorders were a prominent feature in our cohort. Myoclonic movements were observed in 12/15 patients, mostly generalized (10/12) and slow axial (4/10), with 3/12 being stimulus-sensitive and 1/12 persisting during sleep. Dystonia was present in 8/15 patients, with 3/8 having focal dystonia (eg, upper limb, cervical), 3/8 exhibiting segmental dystonia (eg, hemibody or limb involvement), and 2/8 showing generalized dystonia. Parkinsonian syndrome (akinesia and rigidity) was noted in 3/15 patients. Stereotypies were observed in 4/15 patients, including gestural (2/4), oral (1/4, eg, bruxism), and complex gestural-vocal forms (2/4, eg, repetitive hand movements, vocalizations, and hemibody movements).
Clinical Signs on Admission.

Epilepsy was noted in 11 patients and was inaugural in 6 of them. The mean time to epilepsy onset was 29 days [0-180 days]. The different types of seizures were: atonic seizures (5/15), generalized tonic-clonic seizures (5/15), gelastic seizures (3/15), absence seizures (2/15), myoclonic-atonic seizures (2/15), tonic spasms (1/15) or focal clonic seizures (1/15). Status epilepticus occurred in 4 cases. Epilepsy was drug-resistant in 4 cases.
Electroencephalographic Findings
Electroencephalography (EEG) revealed a slowed (n = 8) and poorly organized background rhythm (n = 5). The periodic complexes found in 9 patients were of large amplitude (n = 7), symmetrical (n = 7), diffuse (n = 4), predominantly frontal (n = 3), or posterior (n = 2). The periodic complexes were slow waves (n = 4), spike-and-waves (n = 3), polyspikes (n = 2), or slow waves notched with spikes (n = 2). Periodic complexes were persistent during sleep in 5 patients. Figure 1 shows periodic complexes found in 1 patient.

Electroencephalogram showing periodic complexes with generalized high amplitude slow waves.
Neuroimaging Findings
Brain imaging was normal in 5 cases. Radiological abnormalities observed in 10 patients were classified into 3 patterns: subcortical white matter (WM) lesions, periventricular WM lesions, and brainstem lesions (Figures 2 and 3). All lesions were hyperintense on T2-weighted sequences and hypo- to isointense on T1-weighted sequences, with no evidence of diffusion restriction or contrast enhancement. Subcortical WM lesions were identified in 3 patients. Deep WM lesions, particularly in the periventricular regions, were observed in 4 cases. An association of both patterns was noted in 3 patients. The distribution of these lesions was bilateral and symmetrical in 7 cases. WM lesions were predominantly located in the frontal (3/10), parieto-occipital (3/10), fronto-parietal (3/10), and biparietal (1/10) regions. Two patients had brainstem lesions involving the pons and the middle cerebellar peduncles, presenting a “mustache-like” appearance. The left superior cerebellar peduncle was also affected. Diffuse cortical and subcortical atrophy was noted in 2 patients. Spectroscopy performed in 5 patients showed a choline peak with a decrease in N-acetyl-aspartate in 4 cases and a myoinositol peak in 2 patients. Spinal imaging performed in 1 case, was normal.
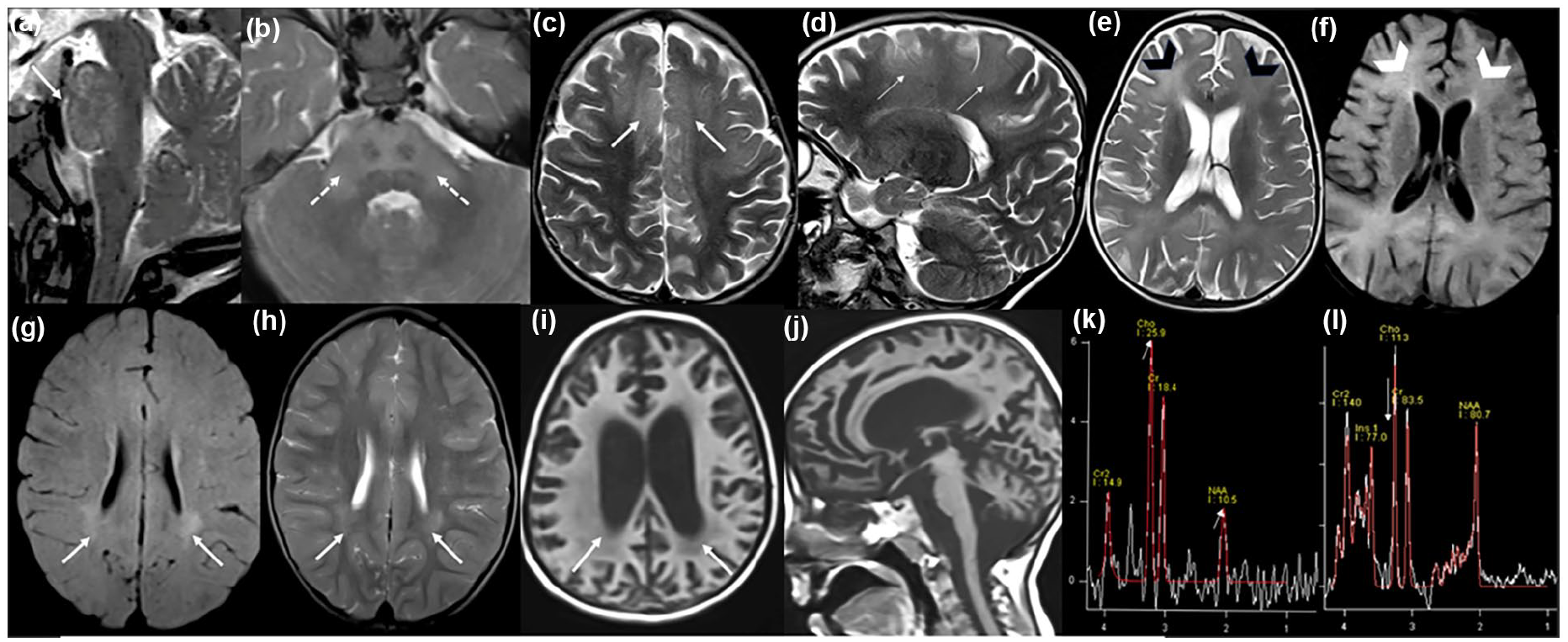
Magnetic resonance imaging of patients with subacute sclerosing panencephalitis. Sagittal and axial T2-weighted images (a and b): T2 hyperintensities in the pons (arrow) and middle cerebellar peduncles, sparing the corticospinal tract and presenting a “mustache-like” appearance (dotted arrows). Axial and sagittal T2-weighted images (c and d): Bilateral subcortical fronto-parietal hyperintensities (arrows). Axial T2-weighted and FLAIR-weighted images (e and f): Bilateral periventricular hyperintensities predominantly in the frontal lobes (arrowheads). Axial FLAIR and T2-weighted images (g and h): Bilateral and symmetrical periventricular hyperintensities predominantly in the posterior lobes (arrows). Axial and sagittal T1-weighted images (i and j): Cortical and subcortical atrophy with periventricular white matter hypointensities (arrows). Spectroscopy of pontine lesions in patient “a” (k): Inversion of the Choline/NAA ratio (arrows). Spectroscopy of periventricular lesions in patient “g” (l): Myoinositol peak (arrow).

Schematic overview of the main radiographic patterns in SSPE.
The “Normal pattern” group, observed in 5 patients, had a mean age of symptom onset at 44 months, with epilepsy present in 4 out of 5 cases (Table 2). Similarly, the “Predominant subcortical lesions” group showed a high incidence of epilepsy, affecting 4 out of 5 patients. In contrast, the “Predominant periventricular lesions” group had a later mean age of onset at 54 months, with epilepsy occurring in 2 out of 3 cases. Notably, the “Predominant brainstem lesions” group, comprising 2 patients, had the earliest mean age of onset at 21 months.
Main Clinical Data Associated with Radiological Patterns in Subacute Sclerosing Panencephalitis.

Lab Findings
Testing for infectious encephalitis was negative, including bacterial culture, polymerase chain reaction for herpes simplex virus, enterovirus, Epstein-Barr virus, COVID-19, and Bartonella antibodies. Testing for autoimmune etiologies was also unrevealing, including Anti-Myelin-oligodendrocyte-glycoprotein (MOG), Anti-N-methyl-D-aspartate (NMDA) receptor antibodies (ab), AMPA- R1 and R2 ab, GABA-B receptor ab, LGi1 ab, and CASPR2 ab.
Lactate dehydrogenase levels were elevated in 9 patients and varied between 252 and 483 UI/l (normal < 240 UI/l).
The study of the CSF revealed normal cytology, positive anti-measles IgG in the CSF in all patients with an elevated index IgG [2,6-7,5]. They all had a multiple oligoclonal bands (OCBs).
Treatment and Evolution
Monthly courses of polyvalent human immunoglobulins were administered to 5 patients; however, the treatment was discontinued after 6 months due to lack of efficacy and high cost. Long-term antiviral treatment with Isoprinosine was given to 4/15 patients. One patient received subcutaneous interferon-alpha (IFN-α) but was lost to follow-up after continuing treatment in another country.
In 4 patients, the initial diagnosis of autoimmune encephalitis led to treatment with intravenous corticosteroids. Clinical improvement in consciousness was noted in 1 patient (P12), though this was transient, followed by secondary deterioration. A transient improvement was also observed in another patient (P5), who received oral corticosteroids for 3 months prior to the diagnosis of SSPE. However, this patient experienced rapid clinical deterioration and died within 6 months of follow-up.
The overall clinical course was marked by progressive neurological decline all cases. Akinetic mutism developed in 7/15 patients, swallowing difficulties in 7/15, dysautonomia in 3/15, and aspiration pneumonia in 2/15 patients. All patients who reached a bedridden state remained clinically stable thereafter and are still being monitored in our consultations, except for 2 patients who were lost to follow-up.
Regarding antiepileptic treatment, the drugs used in our cohort included piracetam (8/11 patients), sodium valproate (7/11), levetiracetam (5/11), clobazam (4/11), and clonazepam (3/11). Seizures were well controlled in the majority of cases; however, 3 patients developed drug-resistant epilepsy.
Discussion
To the best of our knowledge, this study is the first to comprehensively describe the epidemiological, clinical, radiological, therapeutic, and outcome features of SSPE cases following the 2019 measles outbreak in North Africa, offering new insights into the characteristics of SSPE in this post-epidemic context. SSPE is the most severe long-term complication of Measles Virus infection, especially in children who contracted measles in the first year of life. 1 It is a progressive brain disorder caused by persistent wild measles virus. Measles was the second leading cause of infant mortality in Tunisia prior to introduction of measles vaccination in 1979. 7 A World Health Organization expert group reported the global incidence as 4 to 11 SSPE patients per 100 000 measles cases. 8 This incidence is inversely related to measles vaccination coverage. 9 According to the World Health Organization, From 1 January to 30 April 2019, Tunisia experienced a major measles outbreak, with over 3141 reported cases, and 30 deaths, primarily affecting unvaccinated children under 5 years of age. The most affected regions were Kasserine and Sfax, and the outbreak had a high fatality rate of 1%. 10 This large measles outbreak was one of many occurring worldwide, particularly in North Africa. In response, Tunisia revised its vaccination strategy in 2019, with the Ministry of Health launching a nationwide campaign to vaccinate children aged 6 to 11 months, instead of the previous 12-month schedule. 10 There are currently limited epidemiological data on the incidence of SSPE in the post-epidemic period in the Maghreb, particularly in Tunisia. Our department, a referral center for pediatric neurology in northern Tunisia, had not reported any SSPE cases since 2005 until this recent cluster of 15 cases over less than 4 years. This surge, observed specifically in our center, underscores the unusual nature of the outbreak, and further supports the link between the 2019 measles epidemic and the subsequent increase in SSPE cases. However, the lack of comprehensive national epidemiological data highlights the need for broader surveillance to confirm these findings and better understand the true burden of SSPE in the region.
Risk factors that increase the risk of SSPE in children include low socio-economic and educational status, lack of measles vaccination, high number of siblings, and higher birth order. Although we noted a male predominance in our series (12 males vs 3 females) as reported in the literature, there is no evidence of gender-related susceptibility. 11
The onset of SSPE varies from 1 to 10 years after measles infection. In our series, this interval ranged from 1 to 5 years. This shorter delay observed in our series can be attributed to the early age of infection, which was around 6 months on mean. In Tunisia, the measles vaccination age was set at 12 months, leaving a window of susceptibility during the first year of life. Consequently, all our patients were unvaccinated at the time of measles infection, which likely contributed to the early onset of SSPE in our cohort.
Our patients had a typical clinical course of SSPE beginning with mild behavioral disturbances, speech and gait difficulties followed by myoclonic jerks, epileptic seizures, parkinsonism, and at the final stage an akinetic mutism. Among the atypical presentations of SSPE, epilepsy is the most puzzling. It can lead to misdiagnosis when it remains isolated. Seizures were observed in 11 of our 15 patients. This high frequency has not been noted in other series published before 2010, in which epilepsy occurred in 43%-53% of cases of SSPE.12,13 According to the same studies, epilepsy inaugurates the disease in only 23% of cases.12,13 However, in our series, this inaugural aspect was found in almost half the cases (6 patients out of 11). Although rare in SSPE, atonic seizures were the most common type observed in our patients. Similarly, “absences seizures,” found in 2 patients, are exceptional. According to Jović 13 when epilepsy is inaugural, focal seizures are more frequent, whereas generalized seizures are mainly found in late-onset epilepsy. The frequency and puzzling nature of epilepsy, could perhaps suggest that the clinical course of SSPE could vary from one virus variant to another, and from one epidemic to another.
Movement disorders were a prominent feature in our cohort. Epileptic myoclonus remains a core diagnostic feature of SSPE. While slow axial myoclonus is characteristic, we also observed multifocal and generalized forms, consistent with prior reports. 14 Dystonia was identified in 8/15 patients, exceeding the 12.8% prevalence reported in previous studies. 14 Initially focal or unilateral, it can progress to a generalized form, with rare cases evolving into status dystonicus. Similarly, parkinsonism, reported in approximately 6% of SSPE cases, is typically considered a late and infrequent manifestation. 14 However, it may be underrecognized, particularly in pediatric populations where hypokinetic syndromes are less expected. Stereotypies, characterized by repetitive movements or vocalizations, are rare in SSPE (2.6%-8%), 14 but were present in 4 patients in our cohort, manifesting as hand/foot movements, and vocalizations, consistent with findings from the recent systematic review by Garg et al. 14 Other hyperkinetic disorders, such as chorea, tremor, and tics, remain uncommon but have been documented, further highlighting the heterogeneity of movement disorders in SSPE.
Other atypical features such as early age of onset, acute vision loss, pseudotumor cerebri, dysarthria, ataxia, focal deficit, and asymmetric myoclonus, may also lead to a misdiagnosis. 15 These atypical manifestations have been reported in fulminant SSPE. Given their severity and the rapidity of the clinical course, there is no defined stage for these forms.
Given the clinical similarities between autoimmune encephalitis and SSPE, and the differences in their treatment, CSF testing for measles antibody should be considered even in immunized patients prior to the administration of any immunosuppressive therapies. ELISA of CSF for measles virus IgG has a sensitivity of 100%, a specificity of 93.3%. 11 Although not specific, CSF oligoclonal bands are usually positive and the CSF IgG index is typically very high, as observed in our patients. 11 The antibody index that measures the measles-specific IgG fraction in CSF, was not measured in our study, since it is not standard practice. It provides additional specificity, but remains a research tool. 11
EEG in SSPE is characterized by periodic, generalized, bilaterally synchronous and symmetrical high-amplitude slow waves that recur at regular intervals of 5 to 15 seconds called periodic slow-wave complexes. This characteristic electroencephalographic pattern, found in 65% to 83% of individuals with SSPE, 16 was found in more than half of our patients. Sleep EEG is also informative and can be used as a diagnostic tool for suspected cases of SSPE. 17 However, atypical and normal patterns of EEG can also occur.
Although there is no pathognomonic radiological pattern for SSPE, neuroimaging is essential to rule out differential diagnoses. In acute stage, computed tomography and MRI are generally normal. Previous brain MRI studies have shown a predilection for the posterior areas of brain with bilaterally asymmetric signal abnormalities in the parieto-occipital and postero-temporal regions. Later, lesions become more prominent and periventricular white matter, corpus callosum, and basal ganglia can be involved.18,19 A noteworthy radiological aspect observed in our cohort is a propensity for frontal regions, contrasting with the predilection for posterior areas reported previously in the literature. Over time, cerebral atrophy develops invariably and independently from the clinical condition.18,19 Involvement of the brainstem is rarely reported in literature. 20 Only 2 of our patients exhibited brainstem involvement, with pontine lesions extending to the middle cerebellar peduncles, forming a characteristic “mustache-like” pattern. Spectroscopy can reveal a decrease in N-acetyl aspartate and an increase in choline and lactate. This pattern is suggestive of demyelination and inflammation. However, later stages exhibit a diminished N-acetylaspartate-to-creatine ratio, alongside elevated choline-to-creatine and myoinositol-to-creatine ratios. This spectroscopic profile is indicative of gliosis and atrophic processes.
Based on the radiological observations in our cohort, we propose a classification of lesion patterns that may correlate with distinct clinical profiles. However, due to the limited number of patients, no statistical analyses could be performed, and our findings require validation in larger studies. The “Normal pattern” group, despite the absence of visible lesions, showed a high prevalence of epilepsy (4 out of 5 cases), suggesting the potential presence of functional, or microstructural alterations. Similarly, the “Predominant subcortical lesions” group exhibited a high incidence of epilepsy (4 out of 5 patients), supporting the role of subcortical regions in epileptogenesis. Notably, the “Predominant brainstem lesions” group presented with the earliest onset (mean 21 months), likely reflecting the severity of the clinical phenotype associated with extensive brainstem involvement.
Nevertheless, the interpretation of our findings may be limited by the absence of follow-up MRI data, which could have provided a clearer understanding of the evolution of these radiological patterns over time. These preliminary findings highlight the need for larger-scale studies to confirm our observations and further explore the relationship between lesion distribution, symptom onset, and clinical severity.
Treatment and Evolution
Unfortunately, treatment options for SSPE remain limited, and no therapy has proven curative to date. The management of SSPE primarily focuses on supportive care, including the control of seizures and the management of complications such as dysphagia, dysautonomia, and aspiration pneumonia. While many drugs have been proposed to delay disease progression, their efficacy remains variable and often temporary.
Intrathecal interferon-alpha (IFN-α) has emerged as a promising treatment option, with some patients achieving temporary remission and improved quality of life. 21 However, the benefits are often short-lived, and long-term outcomes remain poor, as evidenced by cases where initial remission was followed by severe neurological decline. 21 In our cohort, intrathecal IFN-α was not utilized due to limited availability, reflecting the challenges of accessing advanced therapies in resource-limited settings.
Isoprinosine, both as monotherapy and in combination with ribavirin, has been widely used in SSPE, with some studies reporting stabilization, or slowing of disease progression in certain patients. 22 In our cohort, patients treated with Isoprinosine remained clinically stable during the observation period. However, due to the limited follow-up duration, we cannot yet draw definitive conclusions about its long-term efficacy. The benefits of Isoprinosine remain inconsistent across studies, and robust evidence from clinical trials is lacking. 22 Other antiviral agents, such as oral Lamivudine, Favipiravir and Amantadine, 23 have also been explored, but their effectiveness is limited, and further research is needed to establish their role in SSPE management. 22
Aprepitant, a neurokinin-1 receptor antagonist, has been tested in SSPE but showed no significant benefit except for mild EEG improvement in 1 study. 23 Similarly, immune-based therapies such as rituximab (B-cell depletion) and intravenous immunoglobulins (IVIG) have yielded mixed results. 23 While IVIG showed some benefit in combination with ribavirin in other studies, it was ineffective in our cohort. Rituximab failed to demonstrate efficacy in a single case report. 23 Stem cell transplantation, aimed at immune rebooting, has also failed to show benefit in small case series. 23
Corticosteroids and adrenocorticotrophic hormone (ACTH) have been associated with negative outcomes in SSPE, often leading to rapid disease progression.24,25 In our cohort, patients treated with corticosteroids experienced transient improvements followed by severe deterioration, consistent with previous reports. These findings underscore the importance of avoiding immunosuppressive therapies in SSPE unless autoimmune encephalitis is strongly suspected and confirmed.
The management of myoclonus, a hallmark feature of SSPE, poses significant challenges. Levetiracetam has demonstrated efficacy in controlling myoclonic seizures, while sodium valproate and clonazepam are often used for generalized seizures. 26 Paradoxically, carbamazepine, which is known to exacerbate myoclonic seizures in other forms of epilepsy, has been reported to be effective in treating myoclonus in some SSPE patients. 27 The mechanism underlying this paradoxical effect remains unclear but may be related to the basal ganglia origin of myoclonic activity in SSPE. 27 Despite these interventions, epilepsy in SSPE is frequently drug-resistant, highlighting the need for more effective antiepileptic strategies.
The prognosis of SSPE remains poor, as observed in our cohort. Mortality rates are reported to be as high as 95%, with death typically occurring within 4 years of symptom onset. 28 The average life expectancy for SSPE patients is approximately 3.8 years, ranging from 45 days to 12 years. 29 However, survival can vary significantly, with some patients surviving for several decades. The longest documented follow-up of an SSPE patient is 18 years. 21 This patient, treated with IFN-α using response-based dose adjustments, initially achieved dramatic remission, and improved quality of life for 7 to 8 years. However, the remission was temporary, and the patient later experienced severe deterioration. This case underscores the potential of IFN-α to induce temporary remission but highlights the limitations of current therapies and the urgent need for more effective treatments to improve long-term outcomes. Ultimately, the eradication of measles through effective vaccination programs remains the most beneficial and cost-effective strategy to prevent SSPE and its devastating complications. 30
Conclusion
Our study underlines several key points, notably the importance of diagnosing SSPE in children after the 2019 epidemic. It also highlights the clinical, electrophysiological, biological and radiological features of SSPE in the post-epidemic period. SSPE should be considered in any child presenting with subacute cognitive regression, myoclonus and epilepsy. The history of early measles infection remains an essential key to clinical diagnosis. Periodic complexes are key features in EEG. Subcortical and periventricular white matter demyelination associated with brainstem lesions and progressive atrophy support the diagnosis. Despite effective measles vaccination coverage in our country and in the Maghreb region, a deadly disease like SSPE continues to haunt children. Developing an effective treatment is clearly an urgent concern, as no currently available drug offers a cure for SSPE. Therefore, improving measles vaccination rates and eradicating measles remain the best strategies for controlling and preventing SSPE.
Footnotes
Ethical Considerations
The Ethics Committee of our institution waived the need for ethics approval and patient consent for the collection, analysis and publication of the retrospectively obtained and anonymized data for this non-interventional study.
Consent to Participate
Informed consent was waived by the ethics committee, as the study involved the analysis of existing anonymized data.
Consent for Publication
Consent for publication was waived by the ethics committee, as the study involved the use of anonymized data that posed no risk to the participants’ privacy.
Author Contributions
MBH, ZM, AH, AZ, TBY, MJ, and HBR contributed to the conception of the study. MBH, ZM, ID, and SN were involved in the analysis and interpretation of the clinical and radiological findings. MBH and ZM drafted the manuscript. All authors reviewed, revised, and approved the final version of the manuscript.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.