
Abstract
Several approved Alzheimer’s disease (AD) treatments help manage its associated cognitive symptoms (e.g., donepezil and memantine) or non-cognitive symptoms. However, disease-modifying AD therapies have recently emerged. These treatments aim to slow disease progression by targeting the pathology associated with progressive neurodegeneration. Specifically, two amyloid-targeting therapies (ATTs) are currently approved and available for use in the United States: the monoclonal antibodies donanemab (Kisunla™) and lecanemab (Leqembi®). Both treatments can slow disease progression and cognitive and functional decline in patients with mild cognitive impairment/mild dementia due to AD, but they are associated with class-based safety concerns, notably amyloid-related imaging abnormalities (ARIA). Because advanced practice providers (APPs) such as physician assistants and advanced practice nurses are key to AD patient care, they should be familiar with the biological continuum of AD and with ATTs and understand how to monitor and manage patients receiving these treatments. Therefore, this review aims to educate APPs about these new therapies. Specifically, it summarizes the approved indications and dosing for donanemab and lecanemab, as well as key clinical evidence of efficacy and safety. It also outlines practical considerations around the monitoring and management of patients treated with ATTs, including recommendations about treatment duration, adverse reaction management, and patient counseling.
Keywords
Introduction
Dementia is a clinical syndrome characterized by variable symptoms, including impaired ability to make decisions, process visual and verbal information, form and retain memories, or engage socially (Alzheimer’s Association, 2024; National Collaborating Centre for Mental Health, 2007). Alzheimer’s disease (AD) is the most common form of dementia, accounting for ~60% of cases (Kim et al., 2024). It affects 7 million people in the United States, and its prevalence is expected to rise as the population ages (Alzheimer’s Association, 2024; Rajan et al., 2021).
The pathological cascade in AD is driven by gradual accumulation of amyloid-beta (Aβ) in the brain as both soluble species and eventually insoluble plaques. This leads to the formation of tau aggregates inside neurons, neurodegeneration, and cognitive decline (Agarwal et al., 2023; Yadollahikhales & Rojas, 2023).
AD has a long preclinical phase and is often diagnosed years after cognitive impairment begins (Kim et al., 2024; National Collaborating Centre for Mental Health, 2007). The dementia stage is preceded by mild cognitive impairment (MCI), a stage characterized by measurable but mild symptoms (mainly memory loss) without significant impairment of everyday life (Alzheimer’s Association, 2024; National Collaborating Centre for Mental Health, 2007). MCI may last several years, and it can be confused with “normal” aging (National Collaborating Centre for Mental Health, 2007). Despite certain overlap in declining cognitive performance between both, they are distinct (Gopalakrishnan et al., 2024; Ritchie & Ritchie, 2012).
Advanced practice providers, including physician assistants and advanced practice nurses, are pivotal in dementia care (Alzheimer’s Association, 2024). They are members of the neurology team that deliver patient care in many neurology practices. Their responsibilities include conducting patient examinations, gathering medical histories, writing prescriptions, reviewing diagnostic tests, and managing any adverse reactions occurring during treatment. They also provide a contact point for patients and their care partners throughout the treatment journey. As such, education about eligibility, potential efficacy, and management of potential adverse events is critical.
This review will highlight recent advances in the therapeutic landscape for AD by providing an update on the available options, with a focus on recently approved therapies targeting Aβ aggregates that have demonstrated disease-modifying properties. Specifically, the review will educate on the indications, dosing, efficacy, and safety of this new agent class. Practical considerations around patient monitoring and management as well as patient counseling will also be discussed.
Therapeutic Landscape for AD
Treatments for AD can be divided into symptomatic and disease-modifying treatments.
Symptomatic Treatments
Most currently FDA-approved treatments help manage symptoms of AD; this includes drugs that treat cognitive symptoms and others that treat non-cognitive symptoms.
Acetylcholinesterase inhibitors are used to enhance neurotransmitter availability (Agarwal et al., 2023; Kim et al., 2024). These include donepezil (approved for all stages of AD) as well as galantamine and rivastigmine (both approved for mild-to-moderate dementia due to AD; FDA 1996, 2000, 2001). Additionally, the glutamate receptor antagonist memantine is approved for moderate-to-severe dementia due to AD (FDA, 2003). Of note, cholinesterase inhibitors and glutamate modulators may be combined; an oral combination of donepezil and memantine is approved for moderate-to-severe AD (FDA, 2014b).
On the other hand, treatments that manage non-cognitive symptoms include suvorexant, an orexin receptor antagonist to treat insomnia that has shown clinical efficacy in people with mild-to-moderate AD (FDA, 2014a; Herring et al., 2020), and brexpiprazole, an atypical antipsychotic approved for the treatment of agitation associated with dementia due to AD (FDA, 2023a).
Importantly, while symptomatic treatments provide symptom relief and may have temporary cognitive-enhancing effects, they do not alter the disease course or slow its progression (Agarwal et al., 2023; Cummings et al., 2023a).
Disease-Modifying Treatments
In recent years, there has been a shift toward AD treatments that aim to alter disease progression by targeting the processes underlying the pathology. As discussed below, efforts toward amyloid-targeting treatments (ATTs) that reduce Aβ plaque formation or facilitate its clearance have been relatively successful (Agarwal et al., 2023; Roytman et al., 2023). By contrast, no tau-targeting drugs are currently approved (Cummings et al., 2023b).
Multiple monoclonal antibodies directed against different Aβ forms have been developed (Roytman et al., 2023; Yadollahikhales & Rojas, 2023). Two of these are currently approved and available for use in the US: donanemab (Kisunla™), which targets insoluble N-truncated pyroglutamate Aβ thereby reducing Aβ plaques, and lecanemab (Leqembi®), which targets aggregated soluble and insoluble forms of Aβ (FDA, 2023b, 2024a).
ATTs have demonstrated disease-modifying properties, as they can slow disease progression and cognitive and functional decline in patients with MCI or mild dementia due to AD that have confirmed Aβ pathology. Evidence of their efficacy will be reviewed in the next section. However, slowing of progression does not imply reversal of cognitive/functional deficits, which no treatment has yet achieved. Therefore, ATTs should not be seen as cures; additionally, their use is restricted to patients with brain Aβ accumulation who have MCI or mild dementia due to AD, rather than AD with moderate or severe dementia (Sin et al., 2023).
ATTs have been associated with the risk of amyloid-related imaging abnormalities (ARIA) (observed by magnetic resonance imaging [MRI]) in some ATT-treated patients (FDA, 2023b; 2024a). ARIA can be classified into ARIA-E (“e” for edema, characterized by vasogenic edema or sulcal effusions) or ARIA-H (“h” for hemosiderin deposition, characterized by the leakage of blood degradation products into the brain parenchyma [microhemorrhage] or the subarachnoid space [superficial siderosis]) (Agarwal et al., 2023; Barakos et al., 2022; Hampel et al., 2023; Roytman et al., 2023; Sperling et al., 2011). These events are usually asymptomatic, but symptoms (including rare serious and life-threatening events) may occur (FDA, 2023b, 2024a). Of note, the FDA has placed a boxed safety warning about ARIA in the United States prescribing information of both donanemab and lecanemab (FDA, 2023b, 2024a). Incidence of ARIA as well as other safety concerns associated with these therapies will be discussed below.
Donanemab
Donanemab is a humanized immunoglobulin gamma 1 monoclonal antibody targeting insoluble, N-terminal truncated pyroglutamate Aβ, a modified form of Aβ only present in brain Aβ plaques (FDA, 2024a; Sims et al., 2023).
Indication and Patient Identification
Donanemab is approved for use in MCI or mild dementia due to AD with confirmed Aβ pathology. Presence of Aβ should be confirmed prior to initiating treatment (FDA, 2024a).
In clinical research with donanemab, participants had baseline mini-mental state examination scores of 20 to 28, Aβ pathology (as assessed with florbetapir or florbetaben positron emission tomography [PET]), and tau pathology (as assessed by flortaucipir PET imaging; Sims et al., 2023).
Dosing
Donanemab is administered every 4 weeks as an intravenous infusion over ~30 min. The recommended dose is 700 mg for the first three doses and 1,400 mg subsequently. If an infusion is missed, administration should resume as soon as possible with the same frequency and dose. After dosing, patients should be observed for ≥30 min to monitor for infusion reactions and hypersensitivity reactions. A clinician may consider stopping treatment if PET imaging confirms a reduction of amyloid plaques to minimal levels (FDA, 2024a).
Clinical Efficacy
In the Phase 3 TRAILBLAZER-ALZ 2 trial (NCT04437511), donanemab reduced cognitive and functional decline on the integrated AD Rating Scale (iADRS), a global measure of cognition and function, compared with placebo at week 76 (Sims et al., 2023). Specifically, donanemab slowed disease progression by 35% compared with placebo (based on the iADRS) among patients with low-to-medium tau burden and by 22% in the combined low-to-medium plus high tau burden population. Results controlled for multiple comparisons demonstrated benefit across study populations and outcome measures of cognition and function; significant differences between donanemab and placebo were also reported with the sum of boxes of the Clinical Dementia Rating Scale (CDR-SB), the 13-item cognitive subscale of the Alzheimer’s Disease Assessment Scale, and the mini-mental state examination. Additionally, over 18 months, donanemab reduced the risk of progressing to the next stage of disease versus placebo, as shown by a 37% reduction on the Clinical Dementia Rating global score in the combined population. Further, donanemab delayed disease progression versus placebo by 2.5 months on the iADRS and by 5.4 months on the CDR-SB in the same population (Sims et al., 2023).
Donanemab was switched to placebo at 6, 12, or 18 months for patients who met the treatment completion criteria based on an assessment of Aβ levels via amyloid PET (<11 centiloids on any single scan or 11 to <25 centiloids on two consecutive scans (Sims et al., 2023). MRIs were scheduled at 4, 12, 24, 52, and 76 weeks for amyloid-related image monitoring. Changes in brain Aβ were assessed at 24, 52, and 76 weeks via PET. In the overall population receiving donanemab in the TRAILBLAZER-ALZ 2 trial, 17% of the patients completed treatment at 6 months, 47% at 12 months, and 69% at 18 months (FDA, 2024a). At 76 weeks, brain Aβ plaques decreased by −87 centiloids with donanemab and by −0.7 centiloids with placebo in the combined tau burden population. Aβ clearance at 76 weeks was achieved by 76.4% of donanemab-treated patients (vs. 0.3% with placebo) (Sims et al., 2023).
Clinical Safety
The safety of donanemab has been evaluated in 2,885 patients who received ≥1 dose. The most frequent treatment-emergent adverse events (AEs; affecting ≥10% of patients receiving donanemab) included ARIA-E, ARIA-H microhemorrhage, ARIA-H superficial siderosis, and headache (FDA, 2024a). A total of 13% of donanemab-treated patients discontinued treatment because of an adverse reaction, compared with 4% with placebo (Sims et al., 2023). The most common adverse reactions leading to donanemab discontinuation were infusion-related reactions, which occurred in 4% of patients receiving donanemab versus no patients receiving placebo (FDA, 2024a).
ARIA was observed in 36% of patients receiving donanemab compared with 14% of those receiving placebo. ARIA-E was observed in 24% of patients receiving donanemab versus 2% with placebo, and ARIA-H was observed in 31% of patients receiving donanemab versus 13% with placebo (FDA, 2024a). Symptomatic ARIA occurred in 6% of patients receiving donanemab (Sims et al., 2023). Clinical symptoms associated with ARIA resolved in 85% of those with symptomatic events (FDA, 2024a; Sims et al., 2023). On the other hand, radiographic resolution of the first ARIA-E event occurred in 63% of patients receiving donanemab by 12 weeks, 80% by 20 weeks, and 83% overall after detection (Sims et al., 2023). Intracerebral hemorrhage >1 cm occurred in 0.5% of patients receiving donanemab (including fatal events), compared with 0.2% of those receiving placebo (FDA, 2024a).
Infusion-related reactions occurred in 9% of patients who were treated with donanemab, compared with 0.5% of those receiving placebo. These reactions typically occurred during infusion or within 30 min post-infusion, and 70% occurred during/after the first four infusions. Signs and symptoms included chills, erythema, nausea/vomiting, difficulty breathing/dyspnea, sweating, elevated blood pressure, headache, chest pain, and low blood pressure (FDA, 2024a).
Finally, hypersensitivity reactions, including anaphylaxis and angioedema, occurred in 3% of patients treated with donanemab, compared with 0.7% of those receiving placebo (FDA, 2024a).
Lecanemab
Lecanemab is a humanized immunoglobulin gamma 1 monoclonal antibody targeting aggregated soluble and insoluble forms of Aβ (FDA, 2023b; van Dyck et al., 2023).
Indication and Patient Identification
Lecanemab is approved for use in MCI or mild dementia due to AD with confirmed Aβ pathology. Presence of Aβ should be confirmed prior to initiating lecanemab (FDA, 2023b).
In clinical research with lecanemab, participants had objective impairment in episodic memory as indicated by ≥1 standard deviation below the age-adjusted mean in the Wechsler Memory Scale IV—Logical Memory II. Aβ positivity was determined by PET or CSF measurement of the Aβ1–42 peptide (van Dyck et al., 2023).
Dosing
Lecanemab is administered every 2 weeks as an intravenous infusion over ~1 hr. The recommended dose is 10 mg/kg. If an infusion is missed, the next dose should be administered as soon as possible (FDA, 2023b).
Clinical Efficacy
In the Phase 3 Clarity AD trial (NCT03887455), lecanemab reduced global cognitive and functional decline (as assessed by the CDR-SB) compared with placebo (van Dyck et al., 2023). Worsening in CDR-SB at 18 months was significantly reduced in patients receiving lecanemab compared with those receiving placebo (−0.5 adjusted mean change difference from baseline) (van Dyck et al., 2023); this effect represented a 27% slowing of disease progression (FDA, 2023b). Results were consistent across outcome measures: differences between lecanemab and placebo were also significant using other clinical assessments, such as the 14-item cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-Cog14) and the Alzheimer’s Disease Cooperative Study—Activities of Daily Living Scale for Mild Cognitive Impairment (ADS MCI-ADL). At 18 months, the adjusted mean change from baseline was significantly reduced in patients receiving lecanemab compared with those receiving placebo: 4.1 with lecanemab versus 5.6 with placebo (difference: −1.4) for the ADAS-Cog14, and −3.5 with lecanemab versus −5.5 with placebo (difference: 2.0) for the ADCS MCI-ADL (van Dyck et al., 2023).
At 18 months, the Aβ burden was significantly lower with lecanemab versus placebo: in a PET sub-study, the adjusted mean change from baseline was −55.5 centiloids in lecanemab-treated patients and +3.6 centiloids among those receiving placebo (van Dyck et al., 2023).
Clinical Safety
The safety of lecanemab has been evaluated in 2,090 patients who received ≥1 dose. The most common AEs in Clarity AD (≥10% of in patients receiving lecanemab) included infusion-related reactions, ARIA-H, ARIA-E, and headache. The most common adverse reactions leading to lecanemab discontinuation were infusion-related reactions in the NCT01767311 trial (2% of patients receiving lecanemab versus 1% of patients receiving placebo) and ARIA-H microhemorrhages in the Clarity AD trial (2% of patients receiving lecanemab versus <1% of patients receiving placebo; FDA, 2023b).
ARIA were observed in 21% of patients receiving lecanemab, compared with 9% of those receiving placebo (FDA, 2023b). ARIA-E was observed in 13% of patients receiving lecanemab versus 2% receiving placebo, and ARIA-H was observed in 17% of patients receiving lecanemab versus 9% receiving placebo (FDA, 2023b; van Dyck et al., 2023). Symptomatic ARIA-E occurred in 3% of patients receiving lecanemab (FDA, 2023b; van Dyck et al., 2023). 81% of detected ARIA-E resolved within 4 months of detection (van Dyck et al., 2023). Clinical symptoms resolved in 79% of those with symptomatic ARIA (FDA, 2023b). Intracerebral hemorrhage >1 cm was observed in 0.7% of patients receiving lecanemab (including fatal events), compared with 0.1% of patients on placebo (FDA, 2023b).
Infusion-related reactions occurred in 26% of patients receiving lecanemab, compared with 7% of those receiving placebo; three quarters occurred during the first infusion (FDA, 2023b; van Dyck et al., 2023). Symptoms included fever and flu-like symptoms (chills, generalized aches, feeling shaky, and joint pain), nausea, vomiting, hypotension, hypertension, and oxygen desaturation (FDA, 2023b). Per the appropriate use recommendations developed to guide physicians administering lecanemab, patients should be observed for 3 hr after the first infusion, 2 hr after the second and third infusions, and 30 min after subsequent infusions if no infusion-related reactions have occurred (Cummings et al., 2023a).
Finally, hypersensitivity reactions (including angioedema, bronchospasm, and anaphylaxis) also occurred in patients treated with lecanemab (FDA, 2023b).
Monitoring and Management
Dosing Schedule and Treatment Duration
As discussed, the dosing schedule varies between treatments, with donanemab administered every 4 weeks and lecanemab administered every 2 weeks (FDA, 2023b, 2024a). Importantly, considerations around length of therapy also differ. Treatment with donanemab may be stopped based on an Aβ plaque reduction to minimal levels based on PET imaging (FDA, 2024a). During clinical research, Aβ levels in patients were measured at weeks 24, 52, and 76, and donanemab dosing was stopped (patients receiving donanemab were switched to placebo) based on Aβ levels decreasing below pre-defined PET thresholds: <11 centiloids on a single scan or 11 to <25 centiloids on two consecutive scans (FDA, 2024a; Sims et al., 2023). By contrast, there is no specific guidance on when HCPs should consider stopping treatment with lecanemab (FDA, 2023b).
ATTs and ARIA
To ensure prompt and safe management of any ARIA events that occur, treating HCPs should be familiar with the main risk factors. They also need to recognize and interpret both the MRI changes seen with ARIA and the clinical symptoms suggestive of these events.
Awareness of Risk Factors When Selecting a Treatment
There are multiple ARIA risk factors that should be considered when selecting a treatment. For instance, patients treated with ATTs who are APOE ε4 homozygotes have a higher incidence of ARIA events (including symptomatic and serious events) than heterozygotes and non-carriers (FDA, 2023b, 2024a; Sims et al., 2023; van Dyck et al., 2023). This might be due to ApoE4’s effects in reducing cerebrovascular integrity and increasing neuroinflammation and cerebral amyloid angiopathy (CAA) levels (Foley & Wilcock, 2024). Of note, patients may receive ATTs without APOE genotype testing. However, HCPs should discuss with patients how APOE status affects ARIA risk, with APOE ε4 homozygotes at a higher risk of developing ARIA, and they should recommend APOE genotype testing before initiating treatment (FDA, 2023b, 2024a).
Additionally, many patients with AD have concomitant cerebrovascular disease, including CAA, which is a major cause of intracerebral hemorrhage in older people and a risk factor for ARIA in patients receiving ATTs (Bilodeau et al., 2024; Sveikata et al., 2022).
Antithrombotics might be another potential risk factor. While the available data do not show a substantial increase in ARIA-H among patients receiving ATT with concomitant antithrombotic treatment (FDA, 2023b, 2024a), caution should be exercised when considering a antithrombotic or thrombolytic agent for patients receiving donanemab or lecanemab (FDA, 2023b, 2024a). Of note, however, appropriate use recommendations for lecanemab do highlight that the different risk profile of each drug should be considered; for instance, thrombolytic agents like tissue plasminogen activator should not be used in individuals receiving lecanemab, but standard doses of aspirin or other antiplatelets like clopidogrel may be used in patients who meet other criteria for therapy (Cummings et al., 2023a). If patients require anticoagulants, these recommendations recommend stopping lecanemab dosing until anticoagulants are no longer required (Cummings et al., 2023a).
Radiographic Monitoring
MRI monitoring of patients receiving ATTs is required to detect asymptomatic ARIA, and ad hoc scans are needed to assess for ARIA in patients who develop symptoms consistent with it (Barakos et al., 2022). MRI acquisition protocols should be standardized (Agarwal et al., 2023; Roytman et al., 2023).
ARIA-E can be detected by increased MRI signal on two-dimensional T2 fluid-attenuated inversion recovery (FLAIR) sequences in parenchyma and leptomeninges (Barakos et al., 2022; Sperling et al., 2011). By contrast, ARIA-H is visualized as punctate foci of signal void (areas that are darker than their surroundings) using susceptibility-weighted imaging or gradient recalled echo (GRE)/T2*-weighted MRI sequences (Barakos et al., 2022; Sperling et al., 2011). Unlike ARIA-E, which is more transient and typically resolves radiographically, ARIA-H is irreversible and may stabilize (Agarwal et al., 2023). MRI findings may be used to assess ARIA severity (Table 1; FDA, 2023b, 2024a; Roytman et al., 2023).
ARIA Radiographic Classification (FDA, 2023b, 2024a).
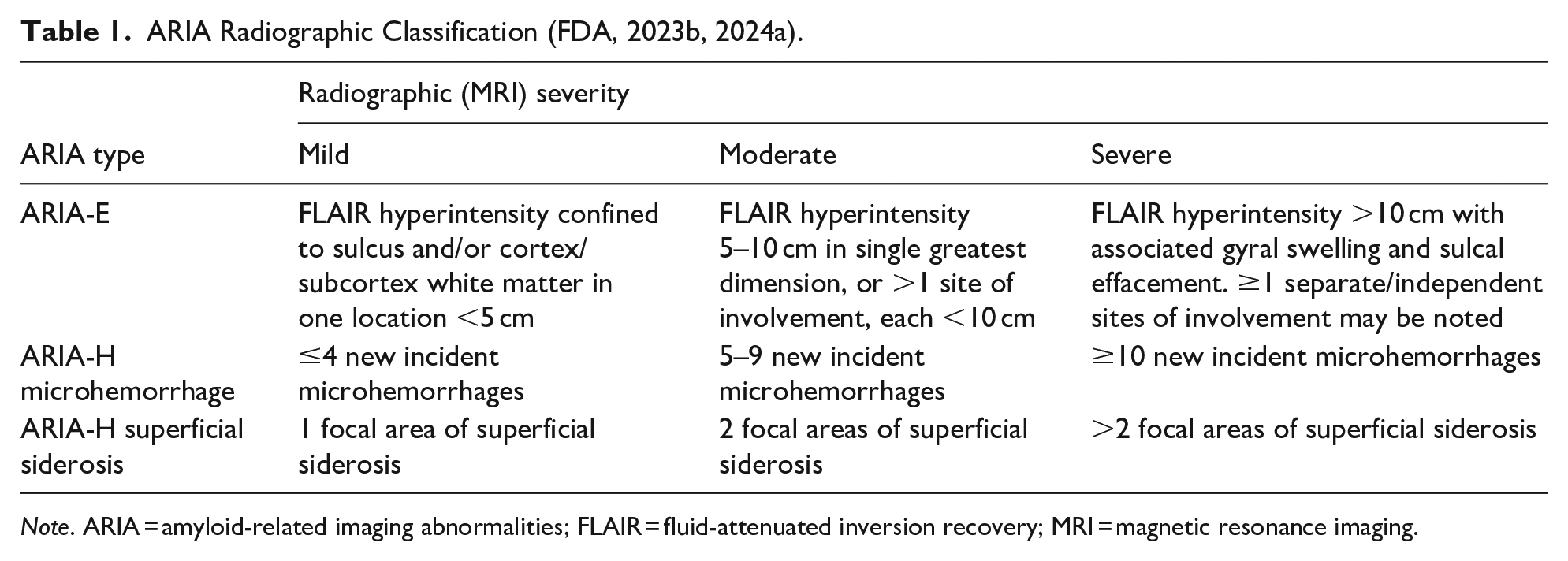
Note. ARIA = amyloid-related imaging abnormalities; FLAIR = fluid-attenuated inversion recovery; MRI = magnetic resonance imaging.
MRI frequency should be based on prescribing information and clinical judgment (Barakos et al., 2022). To monitor for ARIA, a recent baseline MRI should be obtained before ATT treatment starts. Specifically, MRIs should also be done before infusions 2, 3, 4, and 7 with donanemab (FDA, 2024a) or before infusions 5, 7, and 14 with lecanemab (FDA, 2023b). For ARIA-E, most events occurred early during treatment: within three infusions of donanemab (Sims et al., 2023) or 3 months of lecanemab (van Dyck et al., 2023). Therefore, enhanced monitoring is recommended in the early stages of treatment: the first 24 weeks with donanemab (FDA, 2024a) and the first 14 weeks with lecanemab (FDA, 2023b).
Differences Between Clinical Signs of ARIA and Cerebrovascular Events
A history of ATT therapy is an important factor to consider while establishing a diagnosis of ARIA (Roytman et al., 2023). ARIA manifestations resemble lesions seen in CAA, suggesting that AD and CAA may share a common pathway (Sveikata et al., 2022). Indeed, Aβ plays a key role in both: its accumulation in the brain parenchyma leads to AD, but in cerebral vessels it may lead to CAA (Sin et al., 2023; Sveikata et al., 2022).
ARIA symptoms may be confused with ischemic stroke, inflammatory CAA, posterior reversible encephalopathy syndrome, or evolving subacute ischemia (FDA, 2024a; Roytman et al., 2023). Caution is important when patients receiving ATTs present to the emergency department with stroke-like symptoms; HCPs should consider whether such symptoms could be due to ARIA-E before giving thrombolytics to a patient receiving donanemab (FDA, 2024a). Importantly, the window to treat with thrombolytics does not always allow for obtaining an MRI before starting treatment. Therefore, HCPs should assess whether symptoms mimicking stroke may be due to ARIA before administering thrombolytics. Concomitant use of antithrombotic or thrombolytic medications with ATTs may increase the risk of bleeding in the brain (FDA, 2024a).
If there is no evidence of other causes or underlying lesions, ARIA-E should be the first suspicion if parenchymal edema and/or sulcal FLAIR hyperintensity are seen in patients recently treated with ATT (Agarwal et al., 2023). By contrast, prior intracerebral hemorrhage, cerebral microhemorrhage, superficial siderosis, and other lesions such as aneurysm and vascular malformation would be suggestive of CAA (FDA, 2023b, 2024a). Notably, unlike ARIA, CAA is often symptomatic and continues to progress over the course of treatment (Agarwal et al., 2023).
Dose Management
Recommendations for dose management of FDA-approved ATTs in patients with ARIA are provided in Table 2 (ARIA-E) and Table 3 (ARIA-H). This information can be found in section 2 (“Dosing and Administration; Monitoring and Dosing Interruption for Amyloid Related Imaging Abnormalities”) of the corresponding US prescribing information (FDA, 2023b, 2024a).
Dosing Recommendations for Patients With ARIA-E.
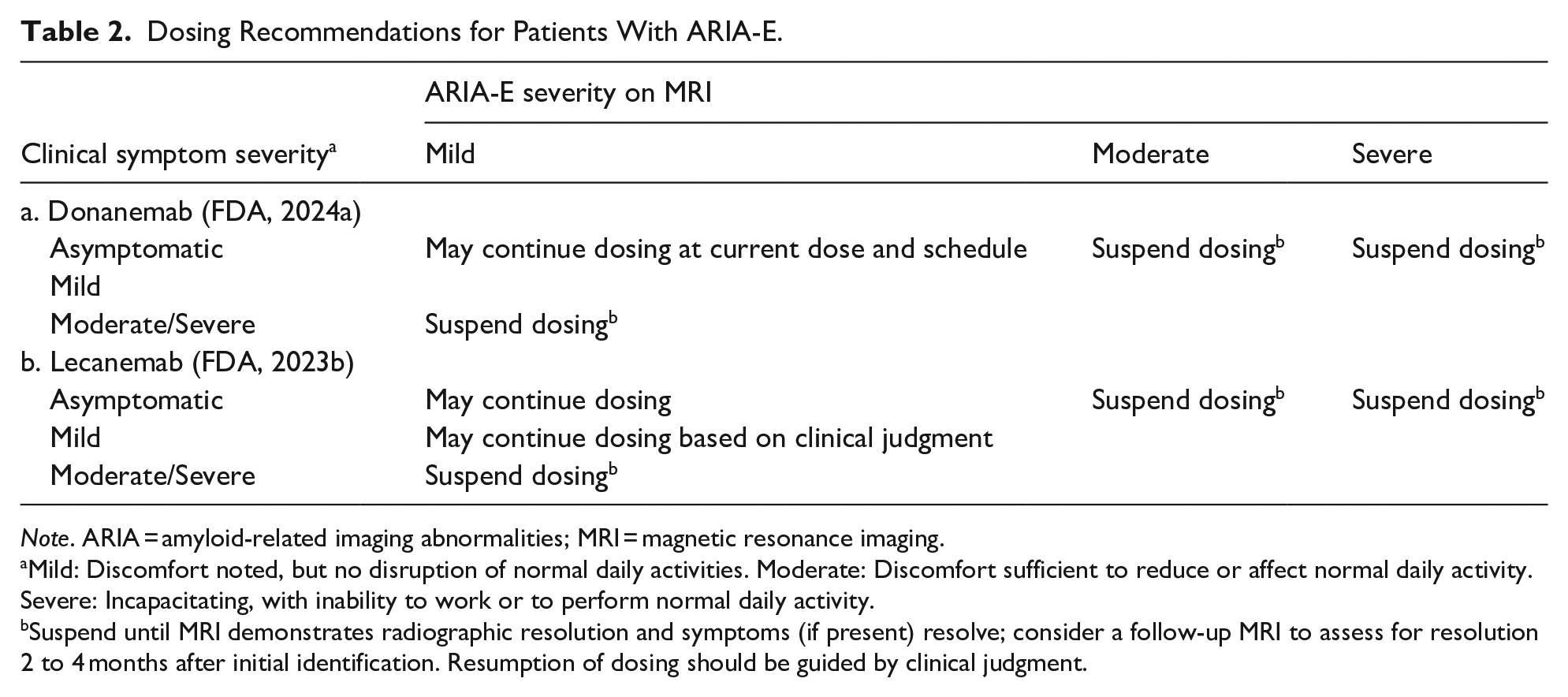
Note. ARIA = amyloid-related imaging abnormalities; MRI = magnetic resonance imaging.
Mild: Discomfort noted, but no disruption of normal daily activities. Moderate: Discomfort sufficient to reduce or affect normal daily activity. Severe: Incapacitating, with inability to work or to perform normal daily activity.
Suspend until MRI demonstrates radiographic resolution and symptoms (if present) resolve; consider a follow-up MRI to assess for resolution 2 to 4 months after initial identification. Resumption of dosing should be guided by clinical judgment.
Dosing Recommendations for Patients With ARIA-H.

Note. ARIA, amyloid-related imaging abnormalities; MRI, magnetic resonance imaging.
Suspend until MRI demonstrates radiographic stabilization and symptoms (if present) resolve; resumption of dosing should be guided by clinical judgment; consider a follow-up MRI to assess for stabilization 2 to 4 months after initial identification.
Suspend until MRI demonstrates radiographic resolution and symptoms (if present) resolve. Use clinical judgment when considering whether to continue treatment or permanently discontinue treatment.
If ARIA are observed on MRI, careful clinical evaluation should be performed before continuing treatment. In patients who develop intracerebral hemorrhage >1 cm during treatment, dosing should be suspended until MRI demonstrates radiographic stabilization and symptoms (if present) resolve. Resumption of treatment should be guided by clinical judgment (FDA, 2023b, 2024a).
Infusion-Related Reactions
If an infusion-related reaction occurs, the infusion rate may be reduced, or the infusion may be discontinued, and appropriate therapy may be initiated. HCPs may consider antihistamines, acetaminophen, or corticosteroids prior to subsequent dosing based on clinical judgment (FDA 2023b, 2024a).
Hypersensitivity Reactions
If any signs or symptoms consistent with a hypersensitivity reaction are observed, the infusion should be discontinued and appropriate therapy should be initiated (FDA, 2023b, 2024a).
Patient Counseling
Communication channels between HCPs and patients receiving ATTs and their care partners must be set up to establish realistic treatment expectations and allow rapid identification of any symptoms. Effective communication should use straightforward language adapted to the background of the patient/care partner and delivered at a reasonable pace, and possibly visual aids (Rentz et al., 2024). It should aim to enhance patients’ comprehension of treatment efficacy and educate them about the scales used to measure effectiveness. For instance, patients/care partners should be aware that ATTs have been shown to slow cognitive and functional decline in the context of large clinical trials, and that this may translate to more time before progressing to the next stage of disease.
Patients should carry information about their ATT treatment (FDA, 2024a, 2024b); this would allow emergency department personnel to be aware of the possibility that any symptoms might be ATT-related. Patients receiving ATTs should also tell HCPs about any other medicines they are taking, particularly antithrombotics (FDA, 2023c, 2024b). Patients should be made aware that some medicines can increase the risk of brain bleeding with ATT treatment (FDA, 2023c, 2024b).
Patients should be informed that established protocols exist to handle any potential adverse events (Rentz et al., 2024). Specifically, advanced practitioners should inform patients that ATT treatment may cause ARIA, which is commonly seen as temporary swelling in the brain that usually resolves over time (FDA, 2023c, 2024b; Rentz et al., 2024). This will help prevent alarm while improving awareness and reporting. They should also inform patients that some people may also have spots of bleeding in or on the surface of the brain; infrequently, bleeding in larger brain areas can occur (FDA, 2023b, 2023c, 2024a, 2024b; Rentz et al., 2024). They should explain to patients that most ARIA events are asymptomatic but serious symptoms can occur. Patients should be informed that they may experience headache, confusion, dizziness, vision changes, nausea, difficulty walking, or seizures; if these occur, patients should be instructed to inform their HCP or go to the nearest emergency department (FDA, 2023c, 2024b). Further, HCPs should counsel patients about the importance of APOE testing, informing them that APOE ε4 carriers have higher risk for ARIA and thus require closer vigilance (Cummings et al., 2023a; FDA, 2023c, 2024b; Rentz et al., 2024).
The conversation should include serious allergic reactions or infusion-related reactions; advanced practitioners should instruct patients to report any relevant symptoms (FDA, 2023c, 2024b; Rentz et al., 2024).
Finally, advanced practitioners should inform patients that they will receive regular MRI scans before and during ATT treatment to monitor for ARIA, and that additional testing may be needed to determine how to treat potential symptoms (FDA, 2023b, 2023c, 2024a, 2024b).
Conclusions
In recent years, disease-modifying treatments for AD that go beyond alleviating symptoms have emerged: two ATTs, donanemab and lecanemab, are currently FDA-approved and available for use in the US for treating AD. Both are indicated for the MCI/mild dementia stages of AD, highlighting the importance of early diagnosis and intervention. Phase 3 results indicate that these treatments can lower the brain Aβ burden and slow cognitive and functional decline, which could extend the period of time during which patients can take care of themselves and thereby reduce the burden on care partners (Vitek et al., 2023). However, when deciding whether to treat a patient with ATTs, the benefit-versus-risk balance needs to be assessed, and adverse reactions need to be carefully considered. Appropriate use of ATTs relies on the ability of advanced practitioners to understand the biological continuum underlying AD, on their familiarity with patient eligibility and with the potential efficacy of ATTs, and on their confidence in recognizing and managing adverse reactions (particularly ARIA, which involves MRI scans). They should also ensure that patients and their care partners understand the potential benefits and harms of ATT, the importance of APOE testing, the possibility of treatment-related reactions and their consequences, and the need for both baseline MRI and monitoring during the course of therapy.
Footnotes
Acknowledgements
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Eli Lilly and Company.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.K., K.V., and B.J. are employees and shareholders of Eli Lilly and Company. CC serves on advisory boards for Eli Lilly and Company, Otsuka, and BrainCheck.