
Abstract
Traumatic brain injury (TBI) and alcohol misuse are major contributors to the global health care burden, both linked to persistent cognitive deficits and mitochondrial dysfunction. This study examined whether post-TBI alcohol exposure exacerbates mitochondrial impairment and contributes to cognitive decline. Adult male Wistar rats were assigned to four groups: Sham, Sham + ethanol (EtOH), TBI, or TBI + EtOH. TBI was induced via lateral fluid percussion 3 days post-craniotomy. Starting 2 days post-injury, EtOH-treated animals received 2.0 g/kg EtOH (i.p.) daily for 5 days. Behavioral testing (Y-maze and open field) was conducted on day 6; brain tissue was collected on day 7 for mitochondrial analysis using the Agilent Seahorse XF Pro Analyzer in the prefrontal cortex (PFC) and retrosplenial cortex (RSC). Rats in the EtOH, TBI, and TBI + EtOH groups showed reduced spontaneous alternation in the Y-maze, suggesting impaired spatial memory. The EtOH group also showed decreased exploratory activity. In the PFC, baseline respiration was unaffected; however, EtOH exposure reduced maximal respiration and glycolytic reserve. In the RSC, TBI and EtOH independently decreased basal respiration, maximal respiration, adenosine triphosphate production, and glycolytic reserve. These findings reveal that alcohol and TBI independently impair mitochondrial bioenergetics in brain regions relevant to cognition and that these deficits parallel behavioral impairments. Disrupted mitochondrial energy metabolism may thus be a key mechanism underlying cognitive dysfunction following TBI and alcohol exposure.
Keywords
Introduction
Traumatic brain injury (TBI) is a leading global cause of disability and death, affecting an estimated 27–69 million individuals each year. 1 In the United States, approximately 611 TBI-related hospitalizations and 190 deaths from TBI occur daily (CDC TBI Surveillance Report 2018–2019), with alcohol involvement reported in 30–50% of cases. 2 Cognitive impairment is a common consequence of moderate-to-severe TBI, affecting up to 43% of patients. 3 Even mild TBIs can result in delayed cognitive recovery, particularly when alcohol is involved.4–7 Pre-clinical studies have demonstrated that binge alcohol exposure prior to TBI worsens functional recovery and disrupts endogenous reparative processes, including impaired sensorimotor recovery and reduced neurogenic responses.8,9 These findings are consistent with clinical observations that many patients are intoxicated at the time of injury.2,10 Work from our group and others has demonstrated that alcohol administration following TBI exacerbates neuroinflammation at the injury site, contributing to worsening cognitive and neurobehavioral outcomes.4,11 Clinical evidence indicates that alcohol consumption during the recovery phase after TBI impairs executive function and hinders rehabilitation outcomes. 12 However, experimental studies addressing the neurobiological and behavioral mechanisms underlying these effects are still limited, particularly with respect to alcohol exposure occurring during the post-injury recovery period.
Emerging evidence suggests that cognitive deficits associated with TBI and alcohol exposure are closely linked to disruptions in cerebral energy metabolism. Brain energy metabolism is critical for neuronal recovery, where increased adenosine triphosphate (ATP) production is required to support the energy demands of cellular repair processes. 13 Mitochondrial respiration is impaired following TBI, 14 and alcohol misuse can reduce mitochondrial oxygen consumption. 15 Both TBI and alcohol misuse contribute to metabolic reprogramming by shifting energy production away from aerobic glycolysis.16,17 Importantly, mitochondrial dysfunction following TBI or alcohol exposure often reflects functional impairments such as altered respiratory control, membrane potential, and coupling efficiency rather than overt loss of mitochondrial proteins. However, the combined impact of TBI and alcohol on brain bioenergetics remains insufficiently understood and warrants further investigation.
Recent findings suggest that the prefrontal cortex (PFC) and retrosplenial cortex (RSC) are particularly vulnerable to the compounding effects of TBI and alcohol. The PFC, which governs executive functions and emotional regulation, is highly sensitive to TBI-related and alcohol-induced cognitive disruption.18–21 Experimental TBI studies demonstrate that synaptic mitochondria in the PFC exhibit significant impairments in state 3 respiration and both complex I- and complex II-driven uncoupled respiration, whereas glial-enriched mitochondrial fractions are often spared. 22 Additional controlled cortical impact studies report marked reductions in cortical mitochondrial respiration, respiratory control ratios, and membrane potential at early post-injury time points, with recovery of oxidative phosphorylation capacity but persistent deficits in membrane potential at later stages, 23 indicating sustained cortical mitochondrial vulnerability even in the absence of ongoing ATP synthesis deficits.
The RSC, essential for spatial memory and contextual processing, is particularly vulnerable to metabolic dysfunction following TBI.24–26 This vulnerability is especially important and may have implications for cognitive dysfunction, given that reduced RSC metabolism has been linked to cognitive deficits in other conditions, such as Alzheimer’s disease.27,28 However, despite its functional importance and connectivity with metabolically vulnerable cortical networks, mitochondrial bioenergetics within the RSC remain largely unexplored in the context of TBI or alcohol exposure. Chronic alcohol use independently increases oxidative stress, alters mitochondrial morphology, and reduces respiratory capacity in these cortical regions,18,29 amplifying the neurobiological consequences of TBI.30,31
Mitochondrial dysfunction is a shared pathological feature of both TBI and chronic alcohol exposure. After TBI, although mitochondrial protein levels may remain unchanged, 32 mitochondrial function is disrupted, leading to impaired ATP production and a secondary energy crisis. 33 This dysfunction drives secondary injury cascades through excess reactive oxygen species, loss of mitochondrial membrane potential, and release of cytochrome c, promoting neuronal death and inflammation.34,35 Alcohol exposure similarly induces altered mitochondrial phenotypes such as mega-mitochondria, reduced respiratory capacity, and disrupted mitochondrial dynamics in the PFC. 18 Alcohol exposure has been shown to impair synaptic mitochondrial function, including reductions in ATP production, membrane potential, and coupling efficiency, while mitochondrial protein levels remain largely unchanged.18,29 Both TBI and alcohol thereby compromise mitochondrial integrity in the cortex, impairing cellular respiration and ATP availability necessary for neuroplasticity and cognitive performance.29,36
Notably, the majority of prior studies assessing brain mitochondrial dysfunction following TBI or alcohol exposure rely on isolated mitochondrial preparations, a methodological approach that may exclude damaged or particularly vulnerable mitochondrial populations during the isolation process. Given the high comorbidity between TBI and alcohol misuse, the present study aimed to examine how post-TBI alcohol exposure influences mitochondrial function and behavioral outcomes using a complementary approach. We employed intact tissue punches from anatomically defined cortical regions to preserve the native cellular and subcellular environment, allowing assessment of mitochondrial bioenergetics within both the PFC and the largely unexplored RSC. Using a lateral fluid percussion (LFP) injury model, we investigated how post-TBI alcohol exposure affects mitochondrial respiration, anxiety-like behavior, and spatial working memory in rats. We hypothesized that alcohol exposure following TBI would exacerbate mitochondrial dysfunction in the PFC and RSC, leading to worsened cognitive and affective deficits
Methods
Animal study design
Adult male Wistar rats (Charles River) arrived at approximately 9 weeks of age (males: 325 ± 50 g; females: 250 ± 50 g) and were acclimated for 1 week. The rats were housed in same-sex pairs under a 12:12-h light/dark cycle and had ad libitum access to food and water. All cohorts were matched for the time and day of testing. Each rat underwent a craniotomy (described below). Three days after craniotomy, animals were randomly assigned to one of four groups: (1) Sham (craniotomy only, n = 23), (2) ethanol (EtOH, n = 22), (3) TBI (n = 21), and (4) TBI + EtOH (n = 21). TBI was induced using LFP to model mild-to-moderate injury, while Sham animals were anesthetized but not injured. On day 2 post-TBI, the EtOH and TBI + EtOH groups received daily intraperitoneal injections of ethanol (2.0 g/kg) for 5 consecutive days, while the Sham and TBI groups were given an equivalent volume of saline.37,38 This regimen was designed to model repeated binge-like ethanol exposure during the subacute post-TBI period. The repeated once-daily dosing schedule was chosen to approximate episodic heavy drinking across consecutive days and to evaluate how repeated intoxication during early recovery influences behavioral outcomes and injury-related pathology.
In our study, ethanol was prepared as a 15% (w/v) solution in sterile saline, filter-sterilized, and equilibrated to room temperature prior to administration. Injections were delivered intraperitoneally using alternating sites and 27–30G needles to minimize local irritation. Throughout the dosing period, animals maintained stable body weight and displayed normal grooming and posture, with no signs of abdominal guarding or piloerection. Routine inspection of the peritoneum revealed no evidence of peritonitis, consistent with previous studies utilizing similar procedures.38,39 This regimen was designed to model binge-level intoxication during the subacute post-TBI period. The 2 g/kg ethanol dose was selected based on established pre-clinical standards known to elicit behavioral and physiological signs of intoxication while maintaining animal welfare.37,38 This dose is commonly used to approximate blood alcohol concentrations observed during binge or heavy episodic drinking in humans. On day 6 post-TBI, 6 h after the final alcohol exposure, the animals underwent behavioral assessments using either the spontaneous alternation behavior test or the open field test (OFT). On day 7 post-TBI, they were euthanized, and tissue samples were collected for further analyses. A full timeline of experimental procedures is presented in Figure 1.
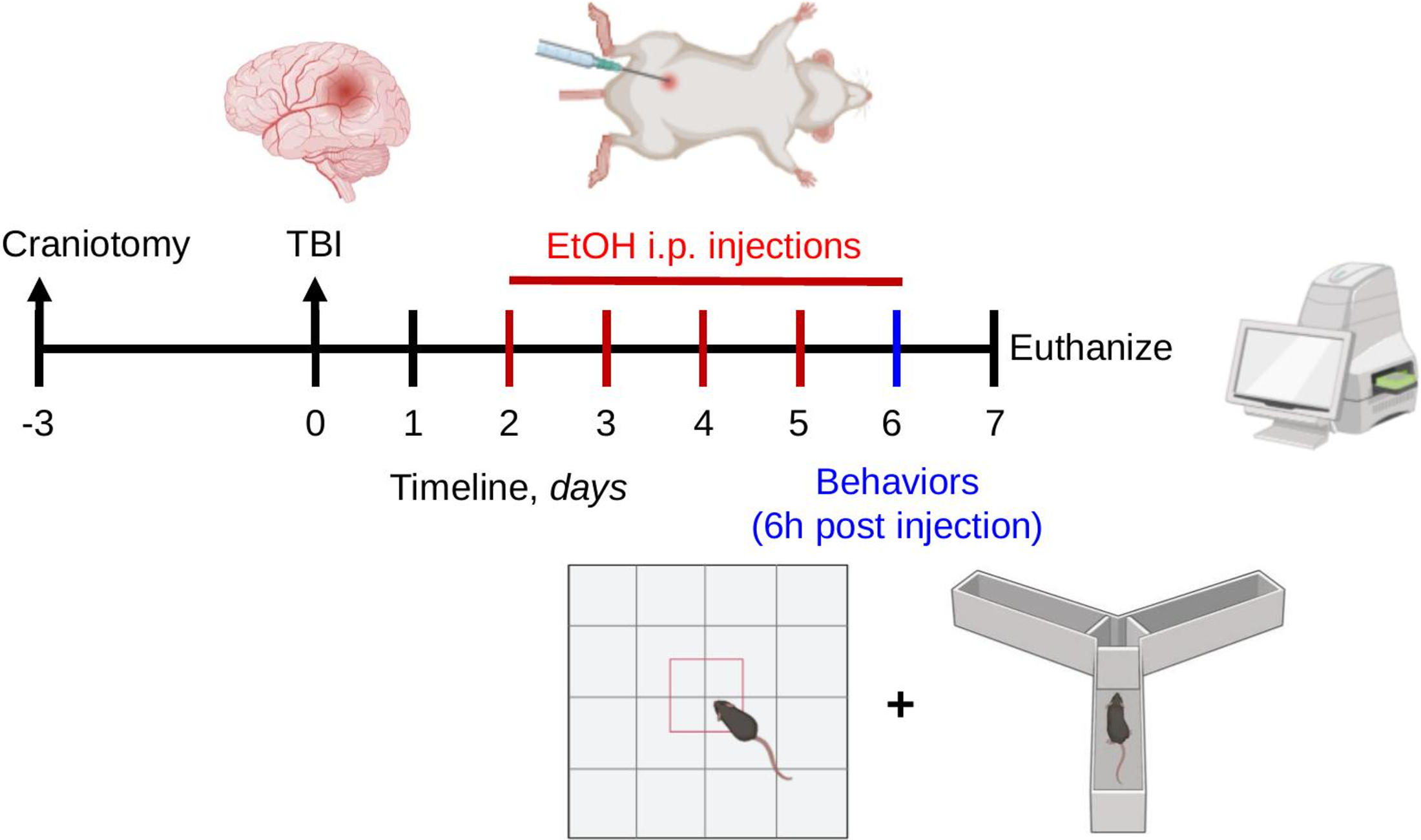
Conceptual framework of experimental design. Rats were randomly assigned to one of four experimental groups: (1) control (craniotomy only, n = 23), (2) ethanol (EtOH, n = 22), (3) traumatic brain injury (TBI, n = 21), and (4) TBI + EtOH (n = 21). All rats underwent a craniotomy, followed by either a sham procedure or TBI using the lateral fluid percussion method 3 days later. Two days after that, rats received daily intraperitoneal injections of ethanol (2.0 g/kg) for 5 consecutive days. On day 6 post-TBI, behavioral assessments, either spontaneous alternation behavior or the open field test, were conducted 6 h after the final alcohol exposure. On day 7 post-TBI, the animals were euthanized, and tissue samples were collected for subsequent analyses. EtOH, ethanol; TBI, traumatic brain injury.
Craniotomy
Three days before TBI, both Sham and TBI animals underwent a craniotomy at coordinates 2 mm posterior to bregma and 3 mm lateral (left) to the midline, as described previously. 39 Animals were anesthetized with 4% isoflurane in oxygen (2 L/min) for 4 min and then placed in a stereotaxic frame (model 900; Kopf Instruments, Tujunga, CA) with continued anesthesia at 2–3% isoflurane in oxygen (1 L/min) administered via a nose mask. After a midline incision, the skull was exposed, and a 3-mm-diameter craniotomy was performed. A Luer Lock connector was placed over the craniotomy and secured with cyanoacrylate glue. Dental cement (Lang Dental Manufacturing, Wheeling, IL) was then applied around the female Luer Lock connector and allowed to harden before the Luer was filled with saline and capped. Topical bupivacaine was applied to the surgical site to provide localized analgesia and minimize post-operative pain.
LFP injury and subsequent alcohol exposure
LFP was performed as published by our laboratory.4,11,39 This model is widely acknowledged in pre-clinical studies for its ability to replicate the histopathological features associated with TBI in humans. Animals were anesthetized with isoflurane (4% induction and 2–3% maintenance) and secured in a stereotaxic frame. The female Luer Lock will be connected to the LFP system (Fluid Percussion Injury, Model 01-B, Custom Design and Fabrication, Virginia Commonwealth University) via pressure tubing. The anesthesia was reduced to 1%, and after 1 min, a mild-to-moderate fluid percussion delivering injury at 28 ± 1 psi (1.905 ± 0.068 atm) was administered. Animals were continuously monitored for signs of apnea, and their respiratory rates and righting reflex times were recorded to assess injury severity. Once all animals regain consciousness, they were returned to their home cages, provided with food and water, and allowed to recover.
Apnea and righting reflex
Following TBI, animals were evaluated for immediate post-injury apnea, the delay between the injury and their first breath. In addition, the righting reflex, the time required for an animal to move from a supine position to a prone position with all four paws touching a flat surface after they were removed from isoflurane, was assessed. This righting reflex models transient unconsciousness. After TBI, the animals were returned to their home cages and provided with food and water.
Spatial working memory assessment
Spatial working memory was assessed using an automated Y-maze apparatus equipped with a camera and computer to monitor movement between arms and time spent in each arm. Distinct spatial cues guided novelty within the maze. Each rat was placed in the maze for a 10-min trial, during which behavior was recorded using a ceiling-mounted video camera and analyzed with ANY-maze software (Stoelting Co.). The maze was made of gray plastic, with three arms (61 × 14 × 35 cm) extending from a central platform at a 120° angle. Each rat was placed in the center of the maze and allowed to move freely among the three arms. An arm entry was defined as the placement of all four paws into an arm, and the sequence of entries was recorded via infrared beams. Alternation was defined as consecutive entries into three different arms on overlapping triplet sets. The percentage of spontaneous alternation is calculated as the ratio of the actual to possible alternations (defined as the total number of arm entries minus 2) multiplied by 100: spontaneous alternation (%) = [(number of alternation)/(total arm entries − 2)]X100. 40 The Y-maze task was performed by an experimenter blinded to the treatment groups.
Open field test
OFT was performed 6 days post-TBI and 6 h after the final ethanol injection, as described previously. 41 The open field apparatus consisted of a square methacrylate chamber (78 × 78 cm) with the floor divided into 25 equal squares (15 × 15 cm). Each rat was placed in the center of the arena and allowed to explore for 10 min while behavior was recorded using a ceiling-mounted video camera and analyzed with ANY-maze software (Stoelting Co.). Anxiety-like behavior was assessed by measuring time spent in the center zone (squares not adjacent to walls) versus the periphery (squares along the walls). The percentage of time spent in the center was calculated as: (center time in seconds/600 seconds) × 100. Additional behavioral metrics included the number of center entries (manually counted; entry defined as all four paws within the center) and total distance traveled. The apparatus was cleaned with Quatricide between sessions to prevent olfactory cues.
Brain slice respiration
Immediately following euthanasia, brains were rapidly removed and sectioned in cold artificial cerebrospinal fluid (aCSF) using a VT1000S vibrating microtome (Leica Biosystems) into 150-µm-thick coronal slices that included the prelimbic subregion of the medial PFC (mPFC) (from +4.56 to 3.72 mm relative to bregma)
42
and the RSC (from −3.14 to 6.30 mm relative to bregma, perilesional site).
43
The aCSF contained (in mM): 120 NaCl, 3.5 KCl, 1.3 CaCl2, 1 MgCl2·6H2O, 0.4 KH2PO4, 5 HEPES, 37 sucrose, and 10
Mitochondrial Stress Test Calculations for Tissue Punches

ATP, adenosine triphosphate; ECAR, extracellular acidification rate; OCR, oxygen consumption rate.
Western blot analysis
Residual cortical tissue from the left hemisphere (the injured side in TBI animals) remaining after tissue slicing for Seahorse experiments was collected, flash-frozen in liquid nitrogen, and stored at −80°C until processing for Western blot analysis. Expression of mitochondrial oxidative phosphorylation (OXPHOS) complexes I–III and V proteins was determined by Western blot analysis (1:1000 dilution, OxPhos Rodent WB Antibody Cocktail, Cat. No.: 45-809-9, and ThermoFisher Scientific) and normalized to the housekeeping protein β-tubulin (1:1000 dilution, beta Tubulin Antibody, Cat. No.: sc-53140, and Santa Cruz Biotechnology).
Statistical analysis
Data are presented as mean ± standard error of the mean. Time-series plots display group means across time points. Sample sizes indicate biological replicates and not technical replicates. Statistical analyses were performed using two-way analysis of variance (ANOVA) to assess the main effects and interactions of EtOH exposure and/or TBI. When significant main or interaction effects were observed, post hoc (unplanned) multiple comparisons were conducted using Dunnett’s test to compare each experimental group to the control group at individual time points. To address focused hypothesis of specific group differences, a priori (planned) multiple comparisons were performed using Dunnett’s test, irrespective of the overall ANOVA outcome. For tissue-specific analyses, unpaired t-tests were used to assess the effects of TBI or EtOH within each tissue. Because data from distinct brain regions were collected and analyzed on separate assays, direct statistical comparisons between tissues were not performed. All statistical analyses were conducted using GraphPad Prism (version 10.2.1).
Results
Apnea and righting reflex post-TBI
To validate the adequacy of the TBI injury and determine if any differences existed between the TBI and TBI + EtOH groups (n = 21–23), post-TBI apnea and the righting reflex were determined. There was no significant difference in apnea duration between the TBI and TBI + EtOH groups (Fig. 2A), with durations of 8.5 ± 1.1 sec and 7.9 ± 1.2 sec, respectively. The righting reflex delay was 202 ± 12.3 sec in Sham animals; 210 ± 7.6 sec in EtOH animals; 424 ± 18.4 sec in TBI animals; and 461 ± 25.3 sec in TBI + EtOH animals (Fig. 2B). Animals in the TBI groups had a significantly longer delay, F(1, 83) = 197.0, p < 0.001, compared to the Sham group. There were no statistically significant differences between the TBI and TBI + EtOH groups.

Apnea duration and righting reflex delay following TBI. Apnea duration
Spatial working memory deficits
Spontaneous alternation (Y-Maze) testing was used to assess both spontaneous alternations and locomotor activity across all experimental groups (n = 9–12). A significant main effect of EtOH was observed to decrease the percentage of spontaneous alternation compared to the Sham group, F(1, 39) = 5.742, p < 0.05; Figure 3A. There was a nonsignificant main effect of TBI and the interaction group, TBI + EtOH, F(1, 39) = 2.949, p = 0.10 and F(1, 39) = 2.813, p = 0.09; respectively, to decrease % spontaneous alternation. However, a priori Dunnett’s tests confirmed significant reductions in % spontaneous alternation for the EtOH (p < 0.01), TBI (p < 0.05), and TBI + EtOH (p < 0.01) groups relative to Sham (Fig. 3A). In addition, the number of entries into the maze was significantly reduced in the EtOH group compared to Sham, F(1, 39) = 12.90, p < 0.001; Figure 3B. Similarly, there was a significant decrease in the number of spontaneous alternations, F(1, 39) = 15.78, p < 0.001; Figure 3C, and in the total distance traveled, F(1, 39) = 15.75, p < 0.001; Figure 3D, in the EtOH groups. While the interaction between TBI and ethanol on spontaneous alternation was not statistically significant, a priori Dunnett’s tests showed that both TBI and TBI + EtOH groups had significantly lower alternation percentages compared to Sham controls.

Spontaneous alternations and distance traveled in the Y-maze during ethanol withdraw post-TBI. Percent
Anxiety-like behavior
The OFT was employed to evaluate exploratory behavior and anxiety-related responses across experimental groups (n = 8–11), providing control for locomotor and affective states that may influence cognitive outcomes. There were no significant differences between any groups for time spent in the inner zone or the number of entries (Fig. 4A and B). However, there was a significant main effect of ethanol to decrease the total distance traveled compared to the Sham group, F(1, 35) = 5.029, p < 0.05; Figure 4C.

Open field testing during ethanol withdraw post-TBI. Time in the inner zone
RSC (perilesional site) tissue respiration
Tissue punches from the RSC of each experimental group were taken (n = 7–8), and a mitochondrial stress test was performed, with representative traces shown in Figure 5A. Basal respiration was significantly decreased in TBI, F(1, 25) = 10.51, p < 0.01, and EtOH, F(1, 25) = 4.681, p < 0.05, with an interaction effect, F(1, 25) = 4.103, p = 0.054, that approached statistical significance (Fig. 5B). We found that maximal respiration was significantly decreased by TBI, F(1, 25) = 14.18, p < 0.001; Figure 5C. In addition, both TBI, F(1, 25) = 4.388, p < 0.05, and ethanol exposure, F(1, 25) = 5.016, p < 0.05, decreased proton leak, although the interaction effect did not reach significance, F(1, 25) = 3.690, p = 0.0662; Figure 5D. TBI also significantly decreased ATP production, F(1, 25) = 8.694, p < 0.01; Figure 7E. Ethanol significantly reduced the glycolytic reserve, F(1, 25) = 6.398, p < 0.05; Figure 5F.
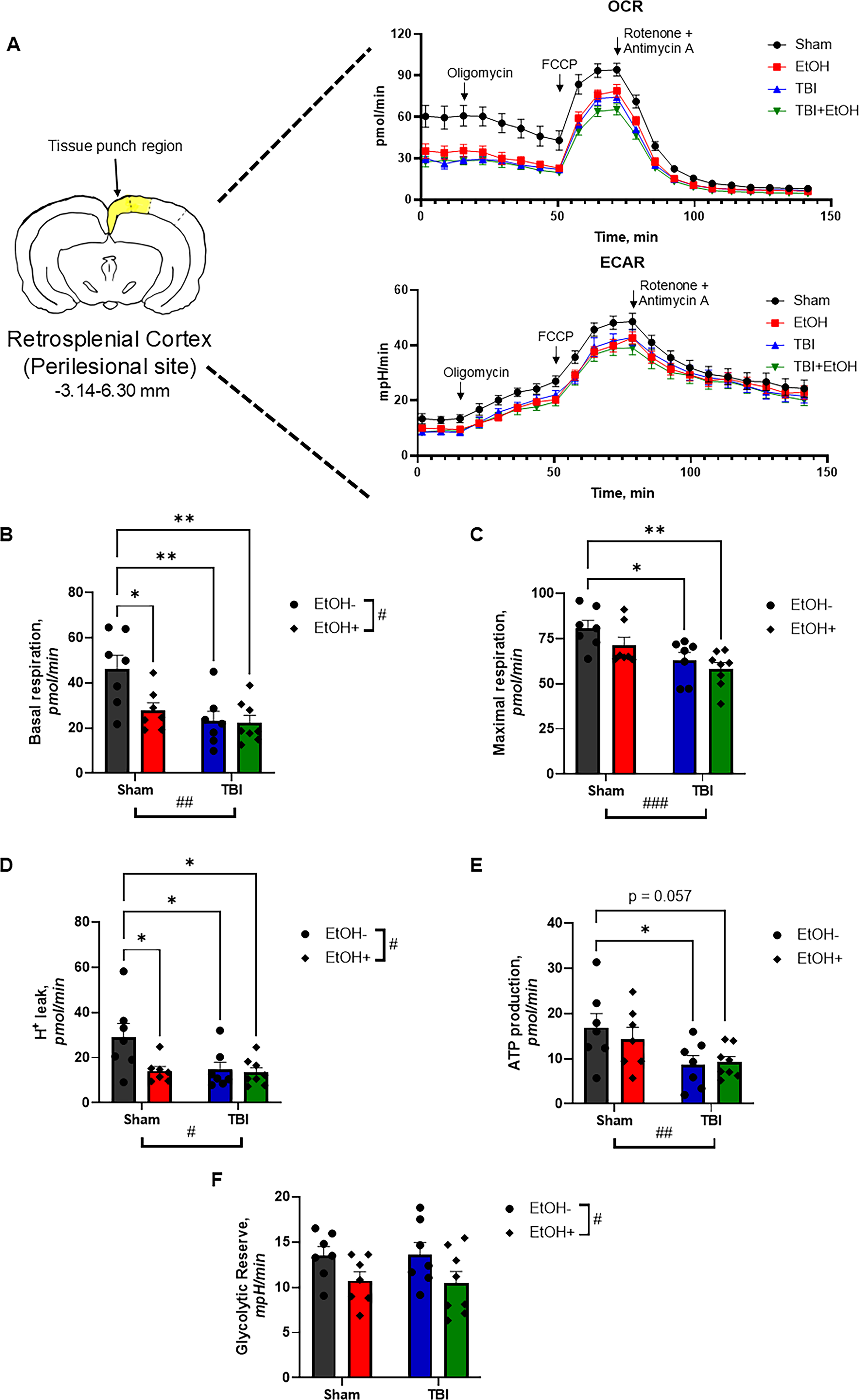
Mitochondrial function and metabolic changes in the RSC following TBI and ethanol exposure. One week after TBI and 1 day following the final ethanol injection, OCR and ECAR were measured from RSC punches. A representative OCR and ECAR trace
To assess whether the observed alterations in mitochondrial respiration were related to changes in the protein expression of electron transport chain (ETC) complexes, we performed Western blot analysis on tissue derived from near the injury site, with a representative blot shown in Figure 6A. Notably, there was a significant decrease in complex I expression in the TBI group compared to the sham group, F(1, 25) = 12.96, p < 0.01; Figure 6B. There were no significant differences in complexes II, III, and V (Fig. 6C–E).
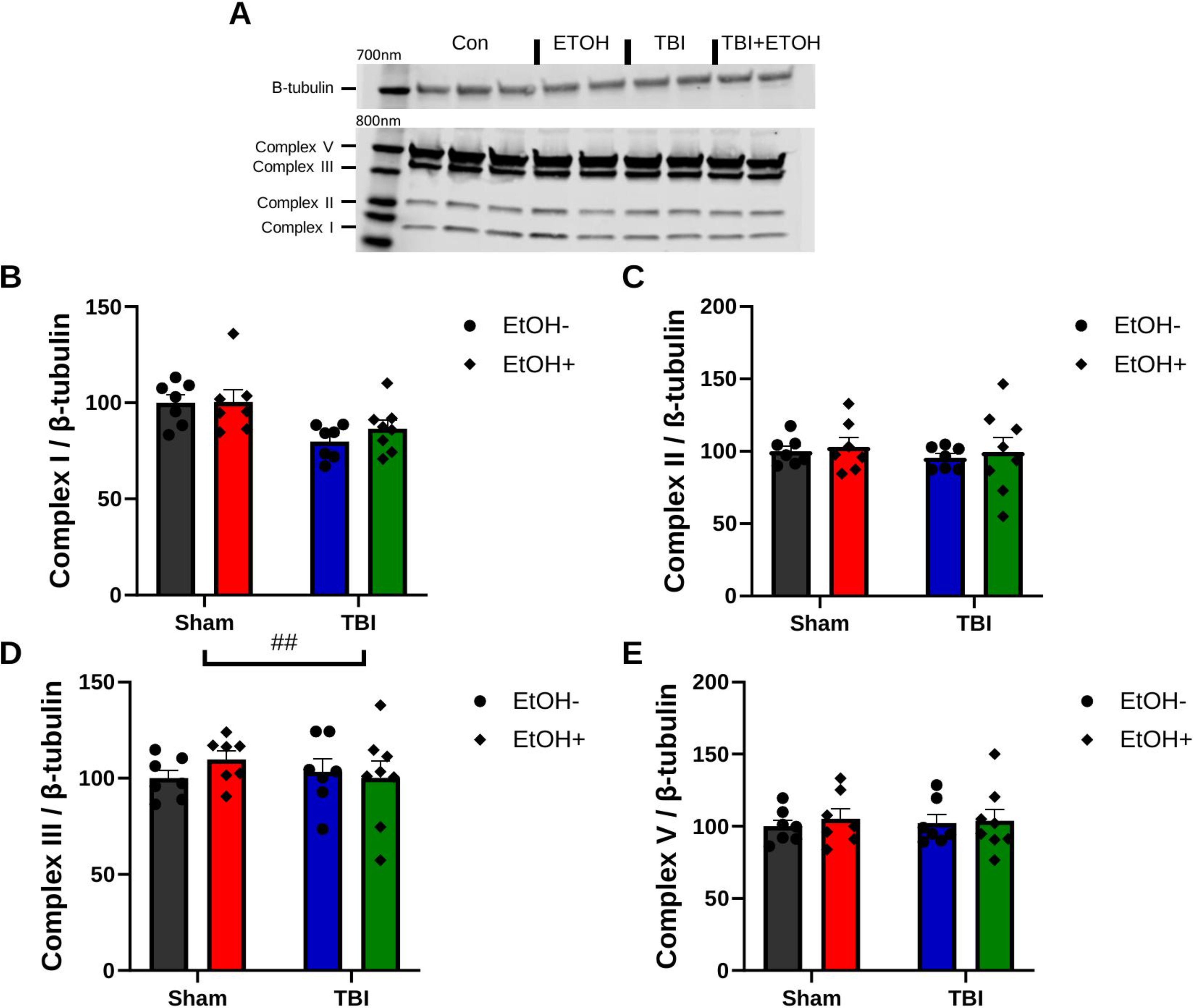
RSC Western blot analysis on mitochondrial ETC complexes. A representative immunoblot showing the expression of mitochondrial ETC complexes I–III and V, with β-tubulin serving as the loading control
PFC tissue respiration
A mitochondrial stress test on PFC tissue punches collected from each experimental group (n = 6–7), with representative traces shown in Figure 7A. Basal respiration was not significantly different between groups (Fig. 7B). However, there was a significant main effect of EtOH on maximal respiration, with EtOH-treated groups showing significantly reduced maximal respiration compared to controls, F(1, 22) = 19.95, p < 0.001; Figure 7C. There were no significant differences in proton leak (Fig. 7D) or ATP production (Fig. 7E), suggesting that the efficiency of oxidative phosphorylation might be preserved despite reduced maximal capacity. Additionally, there was a significant ethanol effect to decrease the glycolytic reserve, F(1, 22) = 19.95, p < 0.01; Figure 7F, suggesting that ethanol exposure limits the ability of cells to compensate for mitochondrial dysfunction by upregulating glycolysis.

Mitochondrial function and metabolic changes in the PFC following TBI and ethanol exposure. One week after TBI and 1 day following the final ethanol injection, OCR and ECAR were measured from PFC punches. A representative OCR and ECAR trace
To determine whether the alterations in tissue punch respiration could be attributed to changes in the protein expression of ETC complexes, OXPHOS protein expression was determined, with a representative blot shown in Figure 8A. There were no significant differences in the expression levels of ETC complexes I, II, III, and V among the experimental groups (Fig. 8B–E), suggesting that the observed functional changes are likely due to modifications in complex activity rather than protein expression.

PFC Western blot analysis on mitochondrial ETC complexes. A representative immunoblot showing the expression of mitochondrial ETC complexes I–III and V, with β-tubulin serving as the loading control
Region-specific effects of TBI and alcohol: Independent impacts on the PFC and injury-proximal site
Given that distinct brain regions may exhibit differential metabolic profiles and responses to injury, data were analyzed by brain region to assess region-specific effects of TBI. Seahorse bioenergetic measures from tissue punches obtained from the PFC and RSC were compared between Sham and TBI groups (n = 6–8 per group; representative OCR and ECAR traces shown in Fig. 9A). TBI significantly reduced basal respiration and maximal respiration in the RSC only (both p < 0.05; Fig. 9B and C), with no significant effects observed in the PFC. Similarly, TBI reduced proton leak and ATP-linked respiration selectively in the RSC, with a trend toward decreased proton leak (p = 0.06) and a significant reduction in ATP-linked respiration (p < 0.05; Fig. 9D and E), while no changes were observed in the PFC. Glycolytic reserve was not significantly altered by TBI in either brain region (Fig. 9F). To evaluate the effects of EtOH, bioenergetic parameters were compared between EtOH− and EtOH+ groups using tissue punches from the PFC and RSC (n = 7–8 per group; representative OCR and ECAR traces shown in Fig. 10A). EtOH produced region-specific effects on mitochondrial respiration, significantly reducing basal respiration in the RSC (p < 0.001; Fig. 10B) with no corresponding effect in the PFC. In contrast, maximal respiration was significantly reduced by EtOH in the PFC (p < 0.01; Fig. 10C), whereas no significant change was observed in the RSC (p = 0.15). EtOH did not significantly alter proton leak or ATP-linked respiration in either brain region (Fig. 10D and E). However, EtOH significantly reduced glycolytic reserve in the PFC (p < 0.001; Fig. 10F), with a trend toward reduction in the RSC (p = 0.07).

Mitochondrial function and metabolic changes in the PFC and RSC following TBI. One week after TBI and 1 day following the final ethanol injection, OCR and ECAR were measured from PFC and RSC punches. A representative OCR and ECAR trace
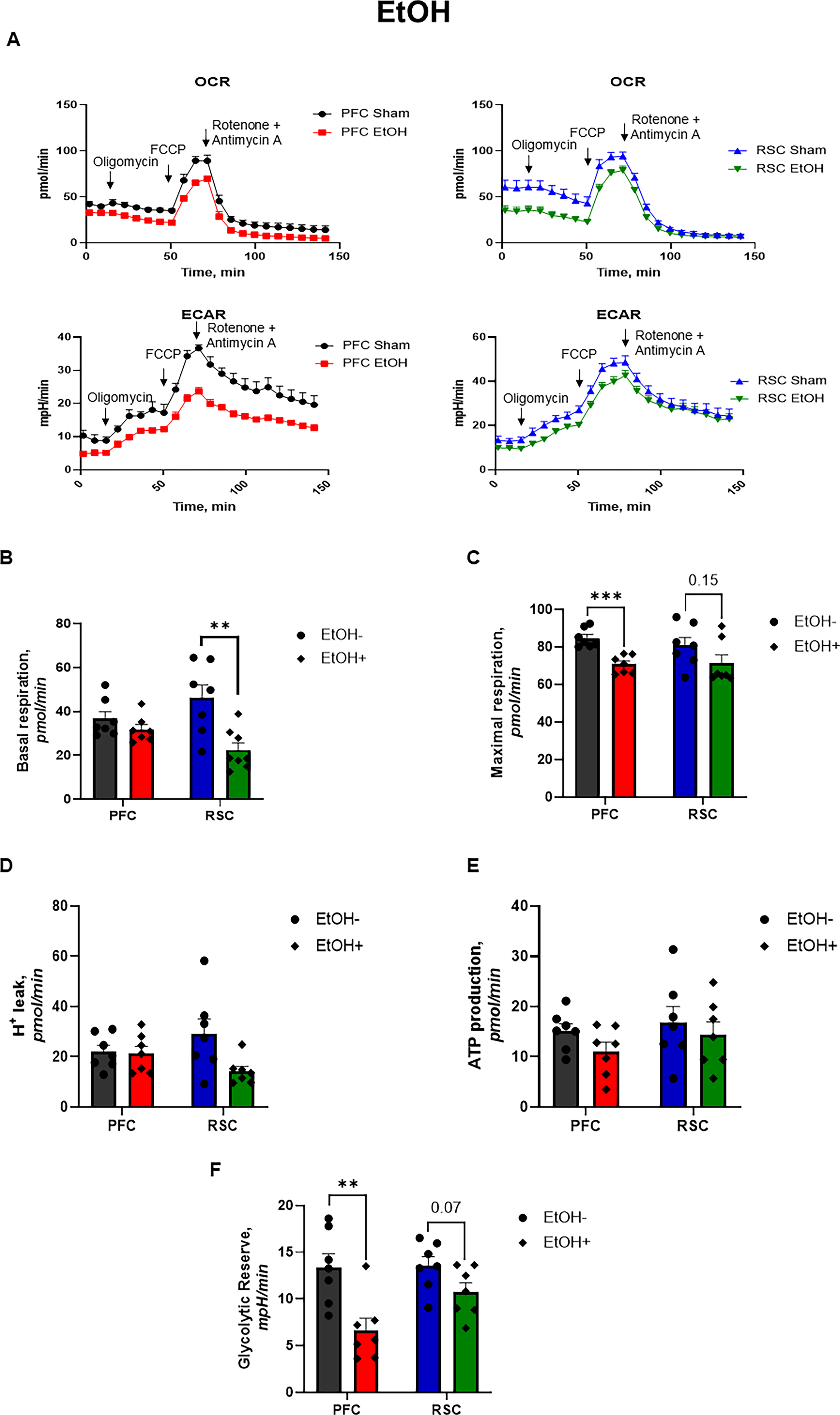
Mitochondrial function and metabolic changes in the PFC and RSC following ethanol exposure. One week after TBI and 1 day following the final ethanol injection, OCR and ECAR were measured from PFC and RSC punches. A representative OCR and ECAR trace
Discussion
This study investigated the effects of mild-to-moderate TBI and ethanol administration on cognitive function and tissue bioenergetics. The results of the study demonstrate that ethanol exposure significantly impairs spatial working memory and exploratory behavior. In addition, both TBI and TBI + EtOH groups had significantly lower spontaneous alternation percentages, indicating significant impairments in spatial working memory. TBI led to marked reductions in basal and maximal respiration, ATP production, and glycolytic reserve in the RSC, indicating localized mitochondrial dysfunction in a region critical for spatial working memory. Ethanol administration impaired mitochondrial function in the RSC, reducing basal respiration, proton leak, and glycolytic reserve. In the PFC, ethanol administration significantly decreased maximal respiration and glycolytic reserve, indicating a compromised capacity to meet increased energy demands. Together, these findings suggest that ethanol exposure and TBI converge on mitochondrial dysfunction within the RSC and are associated with working memory deficits, without evidence of additive effects.
Behavioral implications
Our maze task assessments revealed that the ethanol-administered group exhibited significantly lower percentages of spontaneous alternations and fewer maze entries compared to sham controls. These results indicate impairments in working memory and a reduced exploratory drive, consistent with previous studies documenting similar effects of ethanol on cognitive function.2,4,46 In our experimental conditions, ethanol, TBI, and their combination produced cognitive deficits (Fig. 3A). These observations suggest that ethanol disrupts both memory performance and the motivational processes required for exploration, a dual impact that is especially detrimental during post-injury recovery. We propose that these deficits may stem from the activation of neuroinflammatory pathways, which further impede neural recovery, as previously demonstrated by our group. 4
This impaired recovery may manifest behaviorally as reduced motivation or environmental engagement, consistent with our observation that, despite no changes in conventional anxiety-like measures (Fig. 4A and B), the EtOH group showed a significant reduction in overall exploratory activity (Fig. 3D and 4C). We have previously shown that alcohol does not significantly increase overt anxiety-like behaviors. 41 However, the observed reduction in exploratory activity may reflect underlying neural dysfunction, potentially manifesting as subtle anxiety-like behavior or motivational deficits. Prior investigations have shown that ethanol exposure can lead to deficits in spatial cognition and exploratory behavior, as observed in the current study.47–49 Overall, the reduced exploratory behavior may indicate subtle brain dysfunction caused by ethanol, even if typical anxiety-like behaviors are not affected.
Cellular and mitochondrial bioenergetics
Our findings highlight the critical role of mitochondrial dysfunction in the pathophysiology of cognitive impairments associated with ethanol exposure and TBI. At the tissue level, regions critical for spatial navigation and memory, such as the RSC, exhibited pronounced bioenergetic impairments following ethanol and/or TBI (Fig. 5B–F). Our data show that the exposure to EtOH post TBI does not exacerbate mitochondrial dysfunction beyond the deficits produced by either TBI or EtOH exposure alone. Studies have demonstrated that TBI alone leads to significant reductions in mitochondrial function.50,51 The observed bioenergetics failure in the experimental groups could compromise synaptic plasticity and the energetic integrity of neural circuits, potentially contributing to persistent cognitive impairments as observed in the behavioral assays.52,53 Additionally, we assessed the PFC due to its role in cognition and found that baseline function in the PFC remains relatively unchanged (Fig. 7B). However, ethanol exposure significantly decreased maximal respiration and glycolytic reserve (Fig. 7C and F). This suggests a compromised capacity of neurons to meet increased energy demands during periods of heightened activity, a phenomenon extensively implicated in the development of neurodegenerative processes.54–56 Ethanol’s impact on mitochondrial dynamics is further reinforced by evidence that it disrupts key parameters of energy metabolism, likely through neuromodulatory interactions that lead to subsequent cognitive deficits. 57
Evidence suggests that TBI may increase susceptibility to alcohol misuse, compounding cognitive deficits and posing significant obstacles to neurological recovery.4,11,31 Complementary evidence from the current study indicates significant reductions in maximal respiratory capacity and glycolytic reserve in both the PFC (Fig. 7C and F) and RSC (Fig. 5C and F) following ethanol exposure and TBI. These mitochondrial impairments likely contribute to the observed behavioral deficits, including impaired working memory (Fig. 3A) and diminished exploratory behavior (Fig. 3D and 4C). In particular, the pronounced dysfunction in regions central to spatial memory and navigation offers a mechanistic explanation for the cognitive impairments seen. Moreover, the RSC shares strong functional connectivity with the hippocampus, a relationship critical for spatial memory regulation. 58 Future studies should investigate this network-level interaction by evaluating mitochondrial bioenergetics in the hippocampus, which may further clarify the pathways through which TBI and alcohol exposure disrupt cognitive performance. Taken together, these findings highlight the central role of mitochondrial bioenergetics in supporting cognitive function and highlight the potential therapeutic value of targeting mitochondrial pathways to mitigate the neurodegenerative and cognitive deficits associated with ethanol exposure and TBI.
Region-specific analysis revealed that TBI-induced mitochondrial dysfunction was primarily localized to the RSC, the perilesional site, with significant reductions in basal and maximal respiration, proton leak, and ATP production (Fig. 9B–E). In contrast, ethanol exposure led to broader bioenergetics impairments, affecting both the RSC and the PFC (Fig. 10). These findings suggest that while TBI elicits localized mitochondrial dysfunction, ethanol induces more widespread metabolic disruption. Although we anticipated more pronounced alterations in the PFC following TBI, these effects may have been attenuated by the LFP injury model, which induces mild-to-moderate injury but does not directly target regions such as the PFC (Fig. 9). Our previous work using a weight-drop TBI model reported greater neuroinflammation and tight junction disruption in cortex adjacent to injury, 59 underscoring the importance of injury proximity and severity. Moreover, multiple studies from our group utilizing the LFP model and alcohol vapor exposure have demonstrated robust increases in neuroinflammatory and gliosis markers (GFAP, IBA-1, ED1), whereas alcohol exposure alone does not produce such changes, with elevations observed only in the TBI and TBI + EtOH groups.4,11,60 Additionally, while we hypothesized that the combination of TBI and ethanol would exacerbate mitochondrial dysfunction, the data do not support a synergistic interaction. This may be due to a ceiling effect resulting from the relatively high ethanol dose (2 g/kg), which has been shown to impair bioenergetics independently. 61
Redox and excitotoxic mechanisms underlying mitochondrial dysfunction
The absence of additive effects may also reflect converging mechanisms of injury or a threshold beyond which further mitochondrial compromise is not detectable. Following TBI, excessive and uncontrolled release of glutamate into the extracellular space leads to glutamate overload and subsequent excitotoxicity through overactivation of glutamate receptors.62,63 Elevated extracellular glutamate impairs cystine uptake via system Xc−, resulting in reduced intracellular glutathione levels and promoting oxidative glutamate toxicity-mediated cellular damage. Notably, a mechanistic link between N-methyl-D-aspartate (NMDA) receptor-mediated excitotoxicity and oxidative glutamate toxicity in driving neuronal cell death has been well established. 64 Extensive evidence indicates that mitochondrial bioenergetic impairment arises as a downstream consequence of glutamate-mediated toxicity rather than as a primary insult.65–67 Against the backdrop of glutamate-driven excitotoxicity and glutathione depletion following TBI, alcohol may act as a context-dependent modifier of redox balance and mitochondrial function rather than a simple additive insult.
Alcohol’s effects on glutathione homeostasis and oxidative stress vary markedly with dose, duration, exposure pattern, and withdrawal state, which likely contribute to the heterogeneous findings reported across studies. Acute or low-dose alcohol exposure promotes the generation of reactive free radicals, while its metabolite acetaldehyde directly depletes reduced glutathione. 68 In contrast, chronic or repeated binge alcohol exposure results in sustained glutathione depletion, mitochondrial dysfunction, oxidative stress, and lipid peroxidation, including accumulation of 4-hydroxynonenal, and is accompanied by state-dependent glutamatergic dysregulation within frontal cortical regions such as the anterior cingulate cortex. 69 Importantly, alcohol withdrawal further amplifies glutamatergic hyperexcitability, oxidative stress, and mitochondrial vulnerability, thereby increasing susceptibility to excitotoxic injury.70–72 Within this framework, the lack of additive mitochondrial impairment observed in the combined TBI and alcohol condition may reflect overlapping redox and excitotoxic pathways that converge on shared bioenergetic constraints rather than producing cumulative suppression of mitochondrial respiration detectable at the level of bulk bioenergetic measurements.
Alcohol metabolism itself imposes significant redox pressure within the brain, altering the intracellular balance between oxidative and antioxidant systems. Ethanol is converted to acetaldehyde by catalase and CYP2E1 in the brain, with catalase accounting for the majority of ethanol-oxidizing activity and CYP2E1 contributing to a lesser extent.73–75 Acetaldehyde is further metabolized by mitochondrial aldehyde dehydrogenase 2, yet its excessive accumulation promotes ROS generation, mitochondrial damage, and cell death. 15 Ethanol metabolism increases the NADH/NAD+ ratio, effectively lowering available NAD+, 76 which can enhance superoxide production by respiratory complexes and reduce activity of the NAD+-dependent deacetylase Sirtuin 3 (SIRT3).77,78 Reduced SIRT3 activity leads to hyperacetylation of mitochondrial antioxidant enzymes such as Superoxide dismutase 2 (SOD2), impairing superoxide detoxification and further elevating oxidative stress.79,80 In parallel, alcohol disrupts Ca2+ homeostasis and promotes excitotoxic signaling, 81 processes that are already amplified following TBI. Together, these mechanisms suggest that in the context of TBI-induced metabolic and redox vulnerability, alcohol may exacerbate mitochondrial oxidative stress and impair adaptive metabolic flexibility without necessarily producing additional reductions in tissue respiratory capacity.
In conclusion, our findings demonstrate that both TBI and ethanol exposure independently impair spatial working memory and disrupt mitochondrial bioenergetics in brain regions critical for cognition. Ethanol had a particularly pronounced impact, reducing exploratory behavior and compromising mitochondrial capacity in both the RSC and PFC. These region-specific deficits indicate that mitochondrial dysfunction is a contributor to the cognitive impairments observed after injury and alcohol exposure. TBI alone produced significant mitochondrial dysfunction within the RSC, a region essential for spatial memory, underscoring its direct role in post-injury cognitive decline. Although combined TBI and ethanol exposure did not produce additive suppression of mitochondrial respiration, both insults may converge on pathways that limit bioenergetic capacity and neural resilience. Together, these findings advance our understanding of how metabolic vulnerability contributes to cognitive dysfunction following brain injury and alcohol exposure and highlight mitochondrial bioenergetics as a potential therapeutic target to improve neurological recovery in populations at risk for both conditions.
Future Directions
Future directions include determining whether the absence of additive mitochondrial impairment in the combined TBI and ethanol condition reflects true mechanistic convergence or a bioenergetic ceiling effect. Both insults engage excitotoxic, inflammatory, and redox-sensitive pathways that ultimately compromise mitochondrial function, raising the possibility that they converge on a shared downstream mechanism that reaches a functional threshold beyond which further suppression is not detectable using tissue respirometry. To resolve this, future studies should examine different ethanol dose–response conditions to determine whether additive effects appear when mitochondrial function is not already substantially impaired. Analyses focused on specific cell types, evaluation across acute and withdrawal time points, and more sensitive measures of mitochondrial stress, such as calcium handling, membrane potential, and oxidative damage, are needed to identify interaction effects that may not be detected by respiration measurements alone. In addition, incorporating models of alcohol exposure prior to TBI will be important to determine whether pre-existing metabolic vulnerability alters injury-related mitochondrial outcomes. Together, these approaches will help determine whether the lack of additive effects reflects true convergence on a shared pathway or a ceiling effect and will clarify how TBI and alcohol interact to influence mitochondrial vulnerability.
Limitations
Several limitations of the present study should be acknowledged. First, while the Y-maze spontaneous alternation task was used to assess spatial working memory and exploratory behavior, and the OFT to measure locomotor activity and anxiety-like states, these paradigms are not specific to RSC function. Although spontaneous alternation engages a broader navigation network involving both the RSC and PFC, it does not isolate RSC-dependent processes. Rodents with intact working memory, reflecting normal prefrontal cortical function, typically recall previously visited arms and explore less recently visited ones.82–84 Given the proximity of the injury site to the RSC and the reliance of the task on visual cues, it remains possible that RSC involvement contributed to the observed behavioral outcomes. The RSC is not directly responsible for spontaneous alternation performance per se but plays a critical role in processing allocentric spatial cues that may influence alternation behavior.85–87
Second, more direct assessments of RSC-dependent spatial learning, such as the Barnes maze or Morris water maze, would provide stronger evidence for RSC-mediated cognitive processes. However, these paradigms require extended training periods that could be confounded by injury severity or ethanol exposure. For this reason, we prioritized behavioral assays that did not require pretraining, thereby minimizing potential confounds in post-injury performance. Future studies should incorporate tasks more directly dependent on RSC function to achieve a more comprehensive evaluation of spatial learning and memory. Additionally, assessment of hippocampal integrity would be valuable, as the hippocampus plays a central role in working memory and interacts extensively with the RSC during spatial navigation.58,88,89
Third, due to the limited number of animals available at the time of tissue collection, sampling multiple cortical regions simultaneously was not feasible. As such, our regional analyses were restricted to two cortical areas, limiting conclusions regarding the broader cortical selectivity of TBI- and alcohol-induced effects. Future studies will address this limitation by expanding regional sampling to include additional cortical and subcortical regions, enabling a more complete understanding of injury distribution and specificity.
Fourth, cellular-level resolution was not achievable using bulk tissue punches. Identifying specific cell types affected by TBI and post-injury alcohol exposure would require additional techniques. We are currently developing methods to isolate distinct cell populations from defined brain regions, which will be implemented in future studies to better delineate cell-type-specific responses.
Fifth, RSC and PFC tissue punches were analyzed on separate Seahorse plates rather than on the same plate. As a result, direct plate-matched comparisons of absolute respiration between brain regions were not possible, and no cross-region comparisons were performed. All statistical analyses were therefore restricted to treatment effects within each region.
Last, because this study isolated post-injury alcohol exposure from pre-injury drinking history, the lack of a pre-TBI alcohol component represents an additional limitation. Experimental models have shown that episodic or binge-like alcohol exposure before TBI exacerbates post-injury outcomes, leading to poorer functional recovery and interference with intrinsic repair mechanisms, such as diminished sensorimotor improvement and blunted neurogenic responses.8,9 These preclinical observations parallel clinical reports indicating that a substantial proportion of individuals sustain TBI while acutely intoxicated.2,10 While our findings indicate that post-injury alcohol exposure alone is not more detrimental than TBI itself, these data do not exclude the possibility that pre-injury alcohol exposure, whether chronic or acute, may critically influence injury severity, mitochondrial vulnerability, and long-term functional outcomes. Thus, the changes described here should be interpreted within the context of isolated post-injury exposure, and future studies incorporating both acute and chronic pre-TBI alcohol in the experimental design will be necessary to fully define how alcohol timing and exposure history modify TBI pathophysiology.
Transparency, Rigor, and Reproducibility Summary
The sample sizes were determined a priori using GraphPad Sample Size and Power Analysis online on the Prism Cloud, which indicated that 13 rats per group would provide >80% power to detect both an overall group effect in repeated-measures analysis of variance (primary analysis) and a post hoc difference between the best performing experimental group and controls at p < 0.05 after correction for multiple comparisons. A total of 87 rats were enrolled. Nine were excluded prior to analysis (5 for technical reasons, 3 due to mortality, and 1 for incomplete tissue sampling), leaving 78 animals for outcome assessment. Rats were randomly assigned to experimental groups (Sham, EtOH, TBI, and TBI + EtOH) using a random number generator. All surviving rats received their assigned intervention, and complete behavioral and bioenergetic data were obtained from ≥90% of animals. Three tissue datasets were excluded due to poor quality histological signal.
Investigators responsible for administering ethanol and performing behavioral and bioenergetic assays were blinded to group allocation. Solutions were prepared in identical volumes and appearance to maintain blinding. Ethanol was administered intraperitoneally 6 ± 0.5 h after TBI (2.0 g/kg/day for 5 consecutive days). The LFP injury was delivered at 28 ± 1 psi, and injury severity was confirmed by apnea and righting reflex measures. All reagents were prepared from validated stocks, and batch-to-batch consistency of mitochondrial assay reagents was confirmed by parallel controls. For mitochondrial stress tests, oligomycin A, FCCP, rotenone, and antimycin A were prepared fresh from commercial suppliers and used at standardized concentrations. Normality of behavioral and bioenergetic outcomes was verified using scatter plots and Shapiro–Wilk tests. Replication studies are ongoing and have been preregistered in bioRxiv. All data from this study are upon request to corresponding author.
Authors’ Contributions
X.R.C.-D.: Conceptualization, methodology, investigation, formal analysis, data curation, writing—original draft, and visualization. A.J.-S. and A.M.-S.: Investigation, data curation, and writing—review and editing. L.S., M.S., and S.E.: Conceptualization, data interpretation, and writing—review and editing. P.E.M.: Conceptualization, data interpretation, supervision, writing—review and editing, and funding acquisition.
Footnotes
Author Disclosure Statement
The authors declare that there are no conflicts of interest regarding the publication of this article. No competing financial interests exist. All authors have read and approved the final article and agree to be accountable for all aspects of the work.
Funding Information
This research was supported by National Institute on Alcohol Abuse and Alcoholism grants F32AA031902 (X.R.C.-D.), R01AA025792 (P.E.M.), and T32AA007577 (P.E.M.).