
Abstract
Background and Objectives
Sickle cell anemia (SCA) and glucose-6-phosphate dehydrogenase (G6PD) deficiency are both hereditary diseases of the red blood cells that cause hemolysis. The impact of the interaction of both conditions on the clinical and laboratory presentations of the affected persons is sparse. This study, therefore, correlated G6PD activity with disease severity in persons with SCA by comparing disease severity in G6PD-deficient SCA persons with those with normal G6PD activity.
Methodology
This cross-sectional study was conducted in the department of Haematology and Blood Transfusion of the Obafemi Awolowo University Teaching Hospitals Complex, Ile-Ife, Nigeria. G6PD activity, SCA disease severity, and hematological parameters including reticulocyte counts and Heinz body estimation, bilirubin, and aspartate transaminase were estimated in 67 SCA persons. The results were compared between SCA persons with G6PD deficiency and those with normal enzyme activity.
Results
The prevalence of G6PD deficiency was found to be 23.9%. The G6PD-deficient SCA patients included 4 (25.0%) males and 12 (75.0%) females. G6PD deficiency was significantly higher in females (P = .047). There was no significant difference in disease severity scores between G6PD-deficient and G6PD-nondeficient SCA patients. However, G6PD-deficient persons reported significantly higher episodes of severe vasoocclusive crisis (VOC) per annum (P = .048). The hematological and biochemical parameters were similar between G6PD-deficient and G6PD normal SCA persons except that the G6PD-deficient SCA persons have significantly higher reticulocyte response (P = .001). There was no correlation between disease severity resulting from reduced G6PD activity and Heinz body formation in SCA persons in the steady state.
Conclusion
G6PD deficiency significantly contributes to recurrent painful vasoocclusive crisis in SCA persons in the steady state.
Introduction
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is an X-linked defect that occurs due to mutations in the G6PD genes that cause functional variants with many biochemical and clinical phenotypes. 1 It is the commonest human enzymopathy and it is estimated to affect over 400 million people globally. It is estimated that 15% to 26% of G6PD deficiency occurs in sub-Saharan Africa, causing a significant public health burden.2,3 G6PD deficiency is associated with neonatal jaundice and drug-induced hemolysis. Sickle cell anemia (SCA) is also an inherited disease that causes chronic hemolytic anemia. Its clinical manifestations are due to sickling and destruction of red blood cells leading to anemia. SCA is common throughout most of sub-Saharan Africa, affecting up to 2% to 3% of births in some parts of the continent. 4 In southwestern Nigeria, it has a gene frequency of 2.4%. 4 In the tropics, where malaria and sickle cell disease (SCD) are endemic, there is an unexpected association between SCD and G6PD deficiencies. The coexistence of malaria and G6PD deficiency in a patient with SCA can lead to severe hemolytic anemia, hemoglobinuria, renal failure, and vasoocclusive attacks associated with sepsis. 5 Both sickle hemoglobin gene (HbS) and G6PD deficiency in the heterozygote state confer partial protection against malaria and have, therefore, been selected in geographic areas where malaria is endemic.6,7 The prevalence of both diseases is highest in sub-Saharan Africa, the Arabian Peninsula, and Central India. 8
Studies investigating the potential effects of G6PD deficiency on the severity of SCA have produced conflicting results. Some studies found no effect of G6PD deficiency on the clinical manifestations, hemoglobin levels, or reticulocyte counts of the SCA patient, 9 while others found lower hemoglobin levels in patients having both diseases. 10 However, the impact of the comorbidity of the clinical and laboratory manifestations in patients with SCA has not been fully investigated in our environment. It is anticipated that this understanding may guide recommendations for the screening of SCA patients with G6PD deficiency. This study aimed to determine whether G6PD-deficiency is associated with clinical severity in SCA patients, with the view of recommending early screening and preventive measures to ameliorate the clinical severity.
Methodology
Subjects
This is a cross-sectional study carried out at the haematology department of the Obafemi Awolowo University Teaching Hospitals Complex, Ile-Ife, Nigeria. Participants were adult SCA patients in a steady state. Using the formula for cross-sectional studies,
Clinical Parameters
The severity of the disease was determined using parameters such as the frequency of vasoocclusive crisis (VOC) 13 per year, the number of hospital admissions in the previous 12 months, history of blood transfusion, steady-state hematocrit, white blood cell count, liver and splenic enlargement, the presence of complications such as like chronic leg ulcer, avascular necrosis (AVN), cerebrovascular accident, priapism, gallstones, acute chest syndrome, osteomyelitis, and pneumococcal meningitis. 14 The results of baseline fundoscopy performed on their first presentation were retrieved from their case files. Cardiomegaly was assessed by clinical palpation for the displacement of the apex beat (left ventricular hypertrophy) and left parasternal heave (right ventricular hypertrophy). A tape measure was used to assess liver and/or splenic enlargement below the right or left coastal margins, respectively. The parameters were scored ranging from 0 to 34. The clinical scoring shown in the appendix was adapted from Adegoke and Kuti. 14 A score of ≤7 is regarded as mild, scores from 8 to 17 are regarded as moderate, and >17 is severe.
Blood Sample Collection and Handling
A total of 10 mL of venous blood was collected from each participant according to the standard phlebotomy technique out of which 3 mL of blood was dispensed into bottles containing ethylene diamine tetraacetic acid (EDTA) for full blood count peripheral blood film, reticulocyte count, and Heinz body estimation and 5 mL into anticoagulant-free bottles for measurement of serum total bilirubin and aspartate transaminase (AST) assay and the remaining 2 mL was collected into a separate EDTA bottle for G6PD assay. Full blood count, reticulocyte count, Heinz body estimation, and G6PD assay were analyzed within 6 h of sample collection at 4 °C (not longer than 72 in EDTA bottle at 4 °C). 15 The samples for serum total bilirubin and AST assay were centrifuged at 750g for 15 min to obtain serum. The serum was stored at −20 °C and analyzed in batches for AST, while serum bilirubin was analyzed the same day the samples were collected.
Analysis of Hematological Parameters
Full blood count (FBC) was carried using a Haematology Autoanalyser (AutoAnalyzer [H18] Light) and the results obtained were recorded appropriately. Peripheral blood films were made and stained manually and reticulocyte count, reticulocyte production index, and Heinz body calculations were done manually as previously described. 16
Analysis of Biochemical Parameters
AST was measured spectrophotometrically at 546 nm with a serum normal value of up to 12 IU/L, 17 while total bilirubin was estimated colorimetrically by the methods described by Jendrassik and Grof and the intensity of the color generated by the reaction was proportional to the concentration of the bilirubin. The G6PD deficiency assay was carried out by the Randox enzymatic method18,19 and G6PD activity was categorized using the WHO cut-off of 3 IU per gram of hemoglobin. 20
Statistical Analysis
Data were analyzed with Stata IC version 15 for personal computers. The results were expressed in simple proportions for categorical variables and means and standard deviations for continuous variables. An independent sample t-test was used to compare the means of parameters. Chi-square and Fisher's exact test were used to compare organ complications and grades of disease severity where appropriate. Pearson's correlation coefficient was used to correlate disease severity scores while Spearman rank correlation was used to correlate Heinz bodies with G6PD activity. The level of significance was set as P ˂ .05 at a 95% confidence level.
Results
A total of 69 SCA patients were recruited into the study of whom 2 were excluded because of recent exposure to medications that could have an oxidizing effect on the blood cells and thus provoke hemolysis in G6PD-deficient individuals. Totally, 67 patients were thus analyzed. Table 1 shows the demographic data and medical histories of the SCA subjects investigated. The SCA patients’ ages ranged from 18 to 56 years with a mean and standard deviation of 26.6 ± 8.0 years. The peak age group was 18 to 29 years which accounted for 70.1% of the population. The majority of the patients were females accounting for 52.2% of the population. All had one form of formal education or another. Thirteen (19.4%) reported a history of having severe neonatal jaundice at birth. Fifty-five (82.1%) had a positive history of blood transfusion. The most common indication for blood transfusion was severe anemia which was reported by 53 (96.4%).
Demographic and Medical History of Sickle Cell Anemia (SCA) Patients.

Figure 1 shows the various complications reported in the cohort of SCA patients including cardiomegaly, AVN of bone, retinopathy, leg ulcers, and persistent splenomegaly. The estimated disease severity score ranged from 4 to 11 with a mean (±SD) of 7.04 and 1.86, respectively. Based on the severity score grading system, 56.7% were classified to have mild disease while 43.3% had moderate disease as shown in Figure 2. None were classified as having severe disease.

Sickle cell anemia-related complications in the study population.

Grades of disease severity in sickle cell anemia-related patients.
Table 2 shows the anthropometric, hematological, and biochemical parameters of the patients. Ten (14.9%) SCA patients had packed cell volumes below 20%, while 38.8% had platelet counts above 400 × 109/L. Twenty-five (37.3%) of the SCA patients have suboptimal reticulocyte responses in steady-state (<2.5%). Figure 3 is a quantile normal distribution of G6PD activity in SCA patients which shows that the G6PD activity is normally distributed in the population. However, 16 (23.9%) of the SCA patients were G6PD deficient. Table 3 compares the demography, disease severity, and comorbidity of G6PD-deficient patients and those with normal G6PD status. There was no significant difference in the mean age of patients with G6PD deficiency and those with normal G6PD activity (P = .656). The majority, 11 (68.8%), of the G6PD-deficient patients were less than 30 years of age. There was also no significant difference in the age group between G6PD-deficient and normal SCA patients (P = .628). The G6PD-deficient SCA patients included 4 (25.0%) males and 12 (75.0%) females. There was a significant difference in sex distribution between SCA patients with G6PD deficiency and those with normal G6PD activity (P = .047), with more females being G6PD deficient. There was also no difference in the proportion of G6PD-deficient (68.8%) and normal SCA patients (86.3%) that had a history of blood transfusion (χ2 = 2.54, P = .14). However, G6PD-deficient patients had more severe VOC episodes requiring hospital admission per annum and it was statistically significant (χ2 = 6.08, P = .04). The disease comorbidity indicators were similar in both G6PD-deficient and normal G6PD activity SCA patients (P > .05).
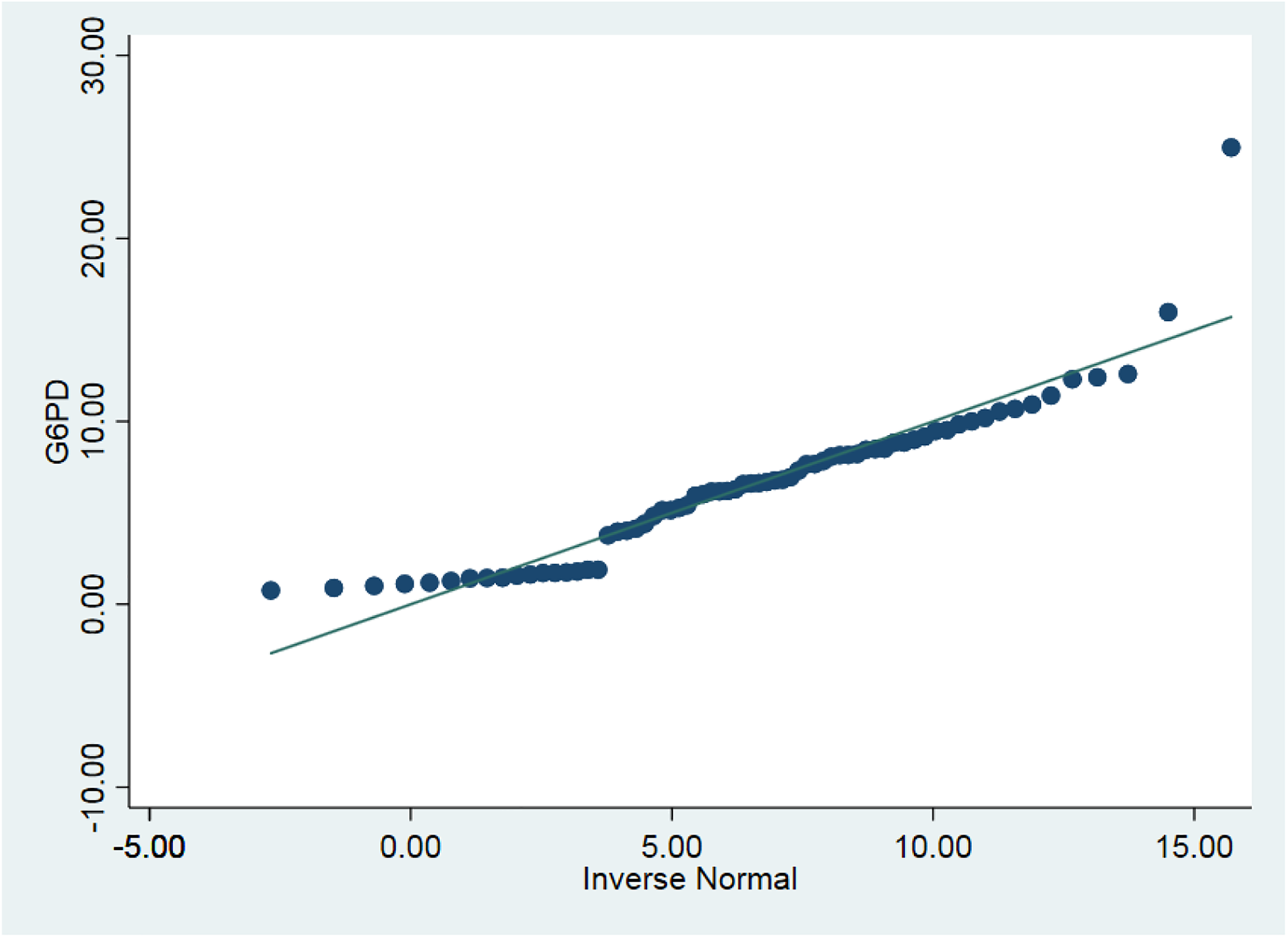
Quartile normal distribution plot of G6PD activity in SCA patients.
Mean Age, Anthropometry, Hematological and Biochemical Parameters of SCA Patients.

Abbreviations: SCA, sickle cell anemia; AST, aspartate transaminase; Hb, hemoglobin; PCV, packed cell volume; RBC, red blood cells; WBC, white blood cells; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RPI, reticulocyte production index.
Comparison of Demographics, Comorbidity, and Disease Severity in G6PD-Deficient and Normal SCA Patients.

Abbreviations: SCA, sickle cell anemia; AVN, avascular necrosis; G6PD, glucose-6-phosphate dehydrogenase; VOC, vasoocclusive crisis.
Figure 4 compares the severity scores between G6PD-deficient and nondeficient SCA patients. The disease severity scores range from 4 to 11 in both G6PD-deficient and nondeficient SCA subjects. The severity scores were higher in G6PD-deficient patients compared to those with normal enzyme activity. However, this was not statistically significant (P = .514). Table 4 shows the hematological and biochemical parameters of G6PD-deficient and nondeficient SCA patients. The hemoglobin levels were lower in G6PD-deficient SCA patients (Figure 5). The G6PD-deficient patients also had a significantly higher reticulocyte response (P = .001) (Figure 6) and a higher reticulocyte production index compared to those with normal activities. Both the total and conjugated bilirubin levels, and more importantly, the unconjugated (indirect) bilirubin levels were higher in G6PD-deficient SCA patients. Similar trends were found with AST which was also elevated in the G6PD-deficient population as shown in Table 4. Heinz bodies were seen in 3 (4.5%) SCA patients. All G6PD-deficient subjects were positive for Heinz bodies. However, there was no correlation between G6PD activity and disease severity scores (r = −0.016 and P = .898) as well as between the presence of Heinz bodies and G6PD activity (r = 0.034 and P = .784).

Comparison of severity scores in G6PD-deficient and nondeficient SCA patients.
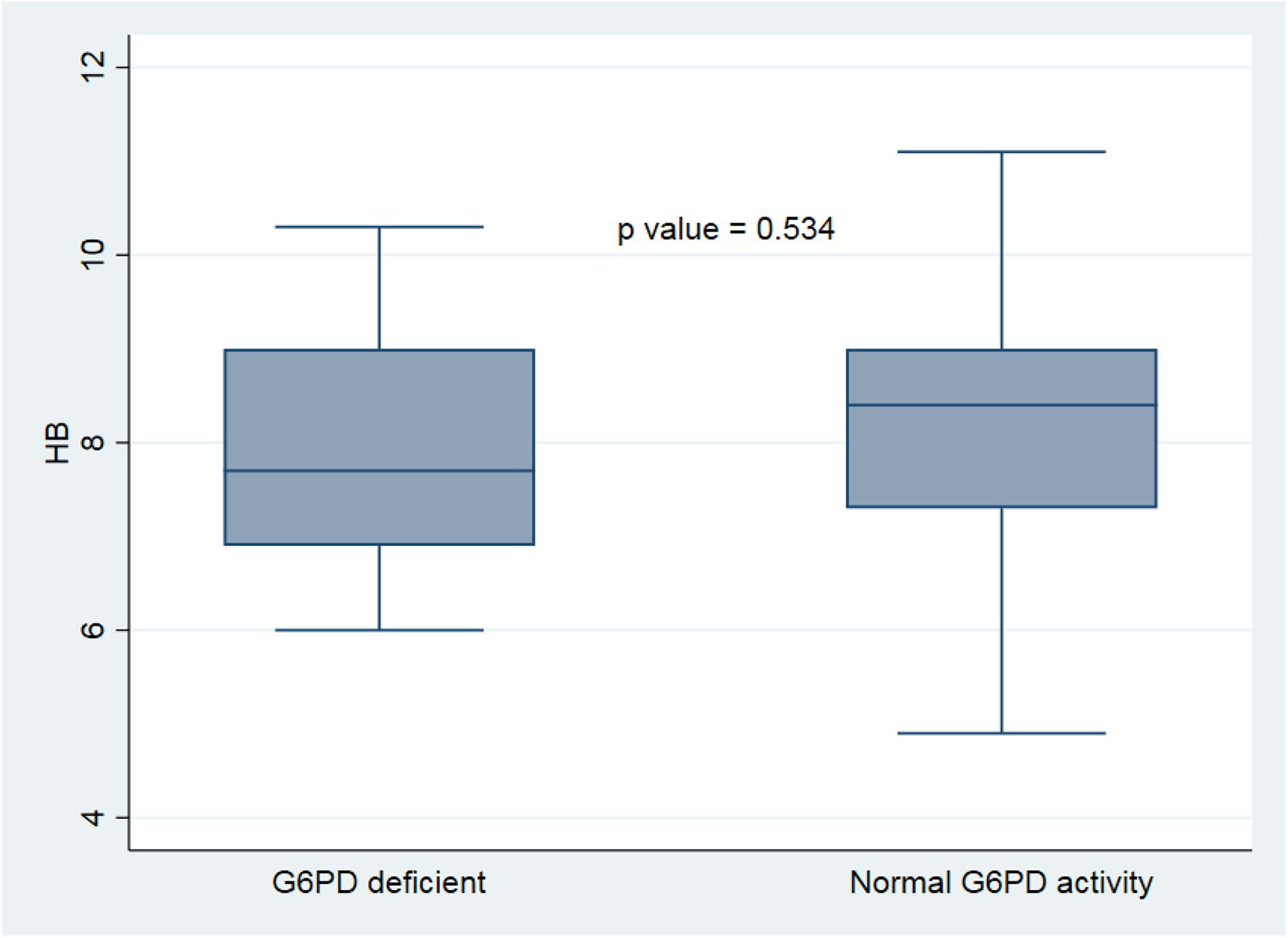
Comparison of Hb levels of G6PD-deficient and normal G6PD SCA patients.

Comparison of the reticulocyte count of G6PD-deficient and normal G6PD SCA patients.
Comparison of Age, Anthropometry, Hematological, and Biochemical Parameters in G6PD-Deficient and Nondeficient SCA Patients.

Abbreviations: SCA, sickle cell anemia; AST, aspartate transaminase; BMI, body-mass index; G6PD, glucose-6-phosphate dehydrogenase; Hb, hemoglobin; PCV, packed cell volume; RBC, red blood cells; WBC, white blood cells; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration; RPI, reticulocyte production index.
Discussion
SCA and G6PD deficiency are both inheritable diseases of the red blood cells that can co-exist in an individual. The impact of comorbidity has been reported in various studies but their findings were not consistent. This study, using estimates of the biochemical activity of red cell enzymes, found the prevalence of 23.9% G6PD deficiency in SCA patients to be within the range found in various national and international studies. From previous studies, the prevalence of G6PD deficiency in SCA patients ranged from 12% to 27.3%.2,21–23 The rates vary with study subject characteristics such as disease state (VOC or steady-state), geographical location, method of estimation, and cut-off level of G6PD activity used to define deficiency state among others. A relatively lower rate was reported by Williams et al, 2 among children less than 15 years old (15.3%) and they observed ethnic variations with SCA patients of Yoruba ethnicity bearing the highest burden of 16.9%. The index study was in the adult population and also conducted in Southwestern Nigeria with predominant Yoruba participants. Bienzle et al 24 also had a relatively lower rate of 16.0% in UCH, Ibadan. However, their study was conducted over 45 years ago and more sensitive methods have emerged over the years that have better performance. It is also necessary to estimate and determine changes in comorbidity over time in our population. International studies among SCA patients in the United States reported lower rates of 13.6%. 25 The latter used genetic assays which are more specific than biochemical assays and the disease burden including SCA and G6PD deficiency was relatively lower in the United States. The prevalence rate found is also similar to the estimated burden ranging from 16% to 25%2,3 of G6PD deficiency reported in the general population. A higher rate was reported by Benkerrou et al 21 among Burkina Faso SCA patients. This also highlights the regional variability in the disease burden. A more recent study in Ghana reported an incidence of 35.8%. 25 Studies that defined deficiency state using cut-off higher than the WHO cut-off of 3IU per gram of hemoglobin tend to report relatively lower prevalence rates. 26
The index study also found no association with age. There was no difference in age between SCA patients with G6PD deficiency and those with normal G6PD activity. The disease conditions are both genetic in etiology and thus the burden may not change significantly with age. The impact of the comorbidity, if significant may alter the life expectancy of persons affected unfavorably; however, this has not been substantiated. Females were found to be significantly more affected than males. This is contrary to the expectation since the G6PD deficiency is X-linked. However, the higher burden on adult females is maybe because of the relatively higher life expectancy of females. A study conducted in early childhood may show a different sex distribution of the disease comorbidity. Nouraie et al 23 reported a predominantly male affection rate; however, their study included SCA patients in the age range of 3 to 20 years. In a similar manner, as the index study, Abubakar et al 22 reported a relatively higher rate of G6PD deficiency among females than males in a study conducted in northern Nigeria. Some other studies have found no difference in the incidence of G6PD deficiency between males and females. 27 However, there is a need for further evaluation of the possibility of change in disease comorbidity in adult SCA patients. The phenomenon whereby females manifest X-linked diseases can be explained by the process of lyonization. The lyonization hypothesis has been used to explain the expression of X-linked disease by heterozygous females. The lyonization hypothesis states that the process of X-chromosome inactivation may be skewed such that the normal X-chromosome may be predominantly inactivated during the process. 28 This may lead to the phenotypic manifestation of a condition by a female with a heterozygous genotype. The phenomenon has been reported to manifest with increasing age. Thus, a significant proportion of adult female SCA subjects may have a clinically detectable G6PD-deficiency state.
The G6PD-deficient SCA patients had significantly more episodes of severe VOCepisodes, necessitating hospital admissions. Individuals with SCA experience a relatively higher background of chronic hemolysis which may result in the higher release of heme into the blood. The release of heme into the blood is known to deplete nitric oxide levels and thus predispose to episodes of vasoocclusive events. 29 Egesie et al observed higher episodes of VOC and hospital admissions in their SCA patients with G6PD deficiency but on the contrary, the subjects in their study had a relatively higher hematocrit. 23 Fasola et al, 26 in a recent study at UCH Ibadan, also reported higher episodes of VOC, hospital admissions, and hepatomegaly in their G6PD-deficient SCA patients. They also noted significantly lower hematocrit in the G6PD-deficient SCA patients. There was no significant difference in organ-specific complications and disease severity scores between G6PD-deficient SCA patients and those with normal activity, although the G6PD-deficient patients had a relatively higher severity score. This is consistent with the reports of Kornberg et al, 18 who noted that the clinical course of SCA with G6PD deficiency was not milder than those with normal enzyme activity. This study found that 14.9% of the SCA patients had hematocrit values below 20%. This is lower than the rates reported by Akinbami et al 30 among SCA patients in Lagos. They reported 31.6% of their study subjects as having a steady-state hematocrit below 20%. Hemoglobin and hematocrit were lower in G6PD-deficient SCA patients. This is consistent with the reports of Simpore et al, 6 Bienzle et al, 19 and Benkerrou et al 21 in their separate studies. The relatively lower hematocrit may be attributed to the slightly accentuated hemolysis in G6PD-deficient SCA patients.6,25,26 Suboptimal reticulocyte response was found in 37.3% of SCA patients in a steady state. The steady state is a state of minimal disease stress and thus it is expected that patients should have an optimal reticulocyte production index for the degree of anemia associated with the disease state. Factors such as micronutrient deficiency, altered iron metabolism, renal impairment, and chronic inflammatory responses are among other factors that have been attributed to the suboptimal reticulocyte response in SCA patients.31–33 Akingbola et al 34 reported in a related study that 43.5% of patients with SCA had suboptimal reticulocyte response in a steady state. The reticulocyte count and reticulocyte production index were higher in the G6PD-deficient patients than those with normal activity. This is anticipated owing to the argument of a relatively accentuated hemolysis in them. Idris et al 35 reported significantly higher reticulocyte count in leprosy patients who are G6PD deficient compared to those with normal enzyme activity. There was no difference in white cell counts, platelet counts, and red cell indices between G6PD-deficient and normal-activity SCA patients in their study. This seems to support the lack of difference in disease manifestation between them and it is consistent with the reports of Simpore et al 6 and Benkarrou et al 21 in their separate studies. Heinz bodies were detected in 4.5% of the study subjects all of whom were G6PD deficient. There was a poor correlation between Heinz bodies and G6PD activity. This is not unexpected as Heinz bodies are particles of oxidized hemoglobin pigments in red cells and are detectable mainly during episodes of intense oxidative stress. The SCD patients were recruited in a steady state when the red cells are least exposed to stress.
The bilirubin and AST levels were found to be increased in SCA patients with G6PD deficiency. There is a paucity of studies comparing AST and bilirubin levels in SCA patients with and without G6PD deficiency. Fasola et al found no significant difference in bilirubin levels between SCA patients with G6PD deficiency and those with normal activity, despite the G6PD-deficient SCA patients having higher bilirubin levels. 23 However, the study is limited by the fact that the assessment of G6PD enzyme activity was not done by either a genetic- or molecular-based study that is more specific and accurate. Possibly patients with acquired deficiency owing to increased oxidative stress may be wrongly classified as G6PD deficient and thus overestimate the burden of G6PD deficiency in the SCA patients.
Conclusions
The study found that severe VOC requiring hospital admissions is significantly higher in G6PD-deficient SCA patients. However, nonevaluation of the alpha thalassemia trait which is a variable that can affect disease severity independent of G6PD activity is a limitation of this study.
Footnotes
Acknowledgments
The authors acknowledge with thanks the consent of our patients to take part in the study.
Authors’ Contributions
The concept of the study was formulated by Salawu; data acquisition, analysis, and manuscript preparation were done by Salawu and Igwilo; manuscript review and approval of the final draft were done by all authors.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Statement of Ethics
The study was approved by the Ethics and Research Committee of the hospital (ERC/2018/01/03).