
Abstract
Genetic analysis of the F8 gene can play a vital role in the diagnosis and management of people with hemophilia A (PWH). Knowing the causative mutation can assist treating clinicians in predicting the risk of inhibitor formation and a person’s response to treatment. It is also a vital tool for genetic counselors in assisting PWH with family planning. Mutational analysis has been limited in developing countries, mainly due to resource constraints; however, with the development of more cost-effective screening tools, these assays are becoming more common in developing countries. Unfortunately, the clinical utility of these assays remain unclear in some of these underresourced countries, resulting in a lack of clinician buy-in. We aimed to highlight the wide-ranging impact mutational analysis potentially has on PWH and their extended families in a developing country.
We used a gel-based in-house method to determine the inversion 22 (Inv22) mutation in a family where a young boy, without a family history, was recently diagnosed with hemophilia A. Inv22 was detected in the proband and his mother, but not in any other direct family member. Therefore, we concluded that the mutation was a de novo mutation in the mother. Subsequently, this result has bearing on at least 23 direct blood-relatives.
Genetic screening extends far wider than the treatment of PWH and is vital in the management of PWH and their extended families. With the availability of cost-effective genetic screening assays this should also be the case in developing countries.
Introduction
Hemophilia A is an X-linked bleeding disorder caused by a deficiency in the blood plasma coagulation protein, factor VIII (FVIII). The inversion of intron 22 (Inv22) of F8 is reported to be the most common causative variation in people with severe hemophilia A. Subsequently, it is recommended that Inv22 detection is performed as the first line of genetic screening in people with hemophilia A. 1 Detection of the causative genetic variation in a person with hemophilia may help predict the possible adverse reactions to treatment, as some variations result in a higher risk for inhibitor development following conventional replacement therapy. 2 Regrettably, hemophilia genetic testing has long been neglected in developing countries, such as South Africa, mainly due to insufficient resources. 3 Fortunately, with implementing a cost-effective conventional polymerase chain reaction (PCR) detection method, Inv22 screening was made more accessible in our resource-restricted setting. With this method, a positive reaction can also be confirmed with Sanger sequencing. In an individual with a positive Inv22, exon 22 of F8 is spliced to an alternative exon to exon 23, exon 23c, thus, clearly distinguishing Inv22 from the wild type. 4 Therefore, our local hemophilia treatment center (HTC) has started to screen all people with hemophilia A in our region for the Inv22 variation. It was previously reported that this variation is under-represented in people with severe hemophilia in our area (∼29%) when compared to the globally reported prevalence (∼45%). In that initial study we screened 62 people (27 families) with hemophilia A, including putative carriers, for the Inv22 variation. 5 Since the publication of our initial study's results and the implementation of Inv22 screening on our diagnostic platform, we have subsequently screened 28 more people with hemophilia (including carriers) from our local HTC. However, securing broader clinician buy-in in our country, especially with genetic carrier detection, with the clinical utility of genetic screening not always apparent in asymptomatic carriers, remains a problem.
With improved management of males with hemophilia and the subsequently increased life expectancy, more males with hemophilia reproduce, increasing the number of potential carriers in the population. Thus, genetic testing may not only assist couples at risk of having offspring with hemophilia to decrease their anxieties regarding reproduction, but also assist clinicians in the management and care of confirmed carriers. It is also important to note that a normal phenotype does not exclude the presence of a causative variation and that genetic testing is required to confirm carrier status. 6 We report a case where the genetic screening influenced a far greater number of individuals than first expected, highlighting the value of genetic carrier screening in our setting. The Health Sciences Research Ethics Committee of the University of the Free State approved this study (UFS-HSD2020/1037/2710). All participants, or the legal guardian in the case of the minor child, gave written informed consent for the publication of their data in this case report.
Case Description
A 3-week-old boy first presented with excessive bleeding at a general practitioner following a frenotomy to correct an ankyloglossia. The bleed was cauterized and he was admitted to the hospital overnight for fluids and observation without any further complications. From roughly 12 weeks of age, minor spontaneous bruising was noticed. At ∼28 weeks of age, the mother noticed excessive bruising around the boy's elbows and took him to his pediatrician. At 30 weeks of age, the proband's FVIII levels were determined at <0.4 IU dL−1, and the subsequent diagnosis of severe hemophilia was made. Consequently, the boy was referred to a pediatric oncologist for a specialist opinion. He received his first FVIII replacement therapy at 31 weeks of age and was subsequently placed on prophylactic Hemlibra® 6 weeks later. The boy is the first child of a then 31-year-old mother, with no family history of bleeding. He was delivered at 41 weeks’ gestation via an uncomplicated cesarean section due to failure to descend. Samples of both the proband and the mother were sent for Inv22 screening. Samples were screened with an in-house developed conventional PCR method. 4 The Inv22 variation was detected in both the proband and the mother. Subsequently, the mother's FVIII level was found to be 20 IU dL−1. It was an important finding, as it is recommended that female carriers with FVIII levels <40 IU dL−1 be considered a person with hemophilia, and treated as such. 7 Consequently, the mother was diagnosed as a woman with mild hemophilia A and referred to a clinical hematologist for management.
We were informed that the mother has 2 younger siblings, a sister (31-year-old) and a brother (27-year-old), neither of whom has children yet. The maternal grandmother has a large family; she is the second of 5 daughters and has 1 younger brother. The maternal grandfather (62-year-old) had a twin sister (deceased) and an older female sibling. There are 8 female and 4 male direct descendants on the maternal grandmother's side of the family. The maternal grandfather's family has 5 female and 5 male direct descendants. He reported that as a youth he had severe pain and swelling in his knees after strenuous running, resulting in impaired function for periods of time. He also has a history of severe joint and back pain, attributed to ankylosing spondylitis. However, with the history of severe and restrictive joint pain, even though unlikely at his age, the possibility of hemophilia was worth investigating in the maternal grandfather to exclude the minor likelihood of an undiagnosed hemophilia. Under diagnosis of inherited bleeding disorders remains a major concern in lower income countries, such as South Africa, with the prevalence in these countries being in most cases <30% than the expected numbers. 8 Therefore, the possibility of a grossly delayed diagnosis in this setting was not necessarily implausible. Furthermore, exceptional cases of somatic mosaicism in asymptomatic maternal grandfathers have been described, resulting in severe hemophilia A in male descendants. Therefore, from a genetic counseling point of view, the risk of mosaicism in maternal grandfathers and grandmothers should be taken into account in seemingly de novo variations. 9 Genetic screening was requested on the family to determine both the possible familial origin of the variation, and the potential presence of the variation in the maternal siblings. With no clinical history of excessive bleeding, it was decided not to screen the maternal uncle unless the maternal grandmother's results indicated it.
The results showed that none of the family members, other than the proband and his mother, had the Inv22 variation (Figure 1). The results were confirmed with Sanger sequencing. Therefore, it was confirmed that the variation was a de novo inversion in the mother. It was previously reported that de novo F8 gene inversions leading to severe hemophilia A occur virtually entirely in male germ cells. 10 Subsequently, evidence shows that most Inv22 variations are of paternal origin, with the male to female ratio reportedly between 15:1 and 300:1. 11 Thus, it is hypothesized that the variation originated during spermatogenetic meiosis of the unaffected maternal grandfather. However, Inv22 variations may also rarely occur during mitotic cell division. 11 Therefore, it was vital to determine whether the maternal aunt had the Inv22 variation. It is especially important because she is of child-bearing age, but without having children, yet. The exclusion of the maternal aunt as a carrier would greatly influence her future family planning and potential bleeding-risk management.
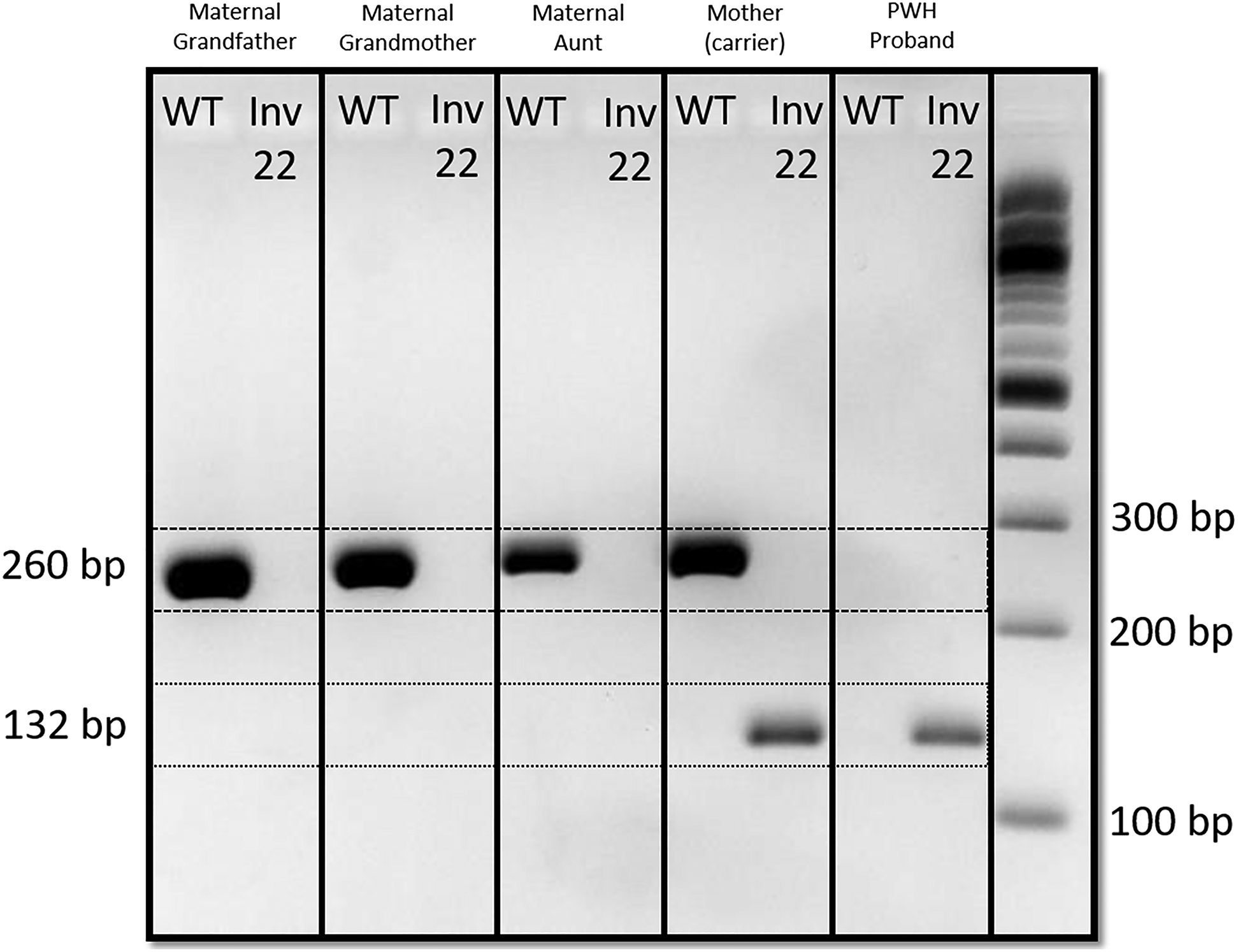
Conventional PCR gel electrophoresis image, depicting the presence of only the 260 bp WT fragment in both the maternal grandparents and the maternal aunt, the presence of both the WT and 132 bp Inv22 inversion fragment in the mother (confirming carrier status), and the presence of only the mutant Inv22 in the affected proband.
Clinical Impact
After closer inspection of the detailed pedigree analysis (Figure 2), it was found that another outcome than the one described could potentially have had a wide-ranging influence on the extended families. For instance, if the causative variation was detected in the maternal grandmother (II:4), all 4 of her sisters (II:5, II:6, II:7, and II:9) could have been potential carriers and her brother a potentially undiagnosed person with hemophilia. It could have possibly resulted in their progeny being affected by hemophilia A. Taken the higher prevalence of female versus male offspring in the maternal grandmother's family, the X-linked pattern of inheritance could potentially have masked the presence of hemophilia A in this family. Therefore, the exclusion of the maternal grandmother as a potential carrier could provide some peace of mind for her extended family.

Pedigree analysis of all ascending blood relatives of the proband (IV:1) up to the great-grandparents. The III:9 indicates the mother of the proband who tested inv22 positive.
Conclusion
We can conclude that this case emphasizes the possible impact of genetic screening on extended families of people with hemophilia A. It is important to note that the effect of genetic screening of families spreads far wider than the medical management of the clinically affected people with hemophilia. Therefore, it should be a core tool in the management of people with hemophilia and their extended families. Furthermore, with the implementation of more cost-effective genetic screening assays, we believe that genetic screening should also become central in the management of people with hemophilia and their families in developing countries. We are also of the opinion that there exists sufficient reasoning behind the establishment of at least one specialized central reference laboratory in under resourced countries, which has the capacity and knowledge to perform comprehensive genetic analysis on people with bleeding disorders. These laboratories should have access to and implement the latest and most informative genetic analysis tools, such as methods that can distinguish between type 1 and type 2 Inv22 variations. However, we do encourage smaller HTCs to perform cost-effective screening assays (such as our novel conventional PCR Inv22 detection assay) at their individual clinics if they have the means to do so. We believe that this will not only result in shorter turnaround times for basic genetic analysis, but also foster greater buy-in from local clinicians and improve the vital interactions between the testing laboratory and treating clinicians.