
Abstract
Our memories are almost magical. We can experience an event for a short moment in time and quickly recall it decades later. This review explores the impact of some relatively new discoveries in the field of flipon biology that provide insight into diseases associated with impaired memory function. I examine how an ancient immune system based on Z-DNA and Z-RNA (collectively called ZNAs) regulates pathways that impact the memories modeled by synapses. The outcomes depend on intracellular defenses activated by endogenous retroelements (ERE) and virus, and on extracellular responses to ZNAs in bacterial biofilms. The bacterial amyloids and complement activation pathways further exacerbate the decline of cognitive and affective functions by inducing remodeling of synapses. In addition to immune EREs, a class of memory EREs potentially acts as ribotransmitters. These RNAs are transported across the synapse to program the connections between neurons that underlie the formation and remodeling of memories. Examples exist of ribotransmitters derived from ERE transcripts and assembled into capsids capable of transsynaptic transmission. In contrast, the immune EREs protect the nervous system by dismantling synapses to prevent viruses and retrotransposons from crossing them. The complexity of the interactions between memory and immune EREs likely give rise to the inverted U-shaped dose-response curves for the therapeutics currently available to treat cognitive decline. Other approaches for disease prevention are suggested, along with those that promote the regeneration and reprogramming of neuronal circuits.
Keywords
Introduction
How does the brain store memories? There are many ways of describing this process: short- versus long-term memory, working versus storage memory, cognitive versus muscle memory, explicit versus implicit, declarative versus nondeclarative; associative versus non-associative; and sense-specific memory. There are various models of memory based on neurons that fire together 1 to holographs that allow its reconstruction. 2 This review will use a very simple model based on a Cantor set to illustrate how complexity can arise from a simple process (Figure 1).3,4 This is not to say that this is how memory is actually implemented, but it illustrates the processes we will discuss. The Cantor set starts with a line divided into thirds from which the middle third is removed to generate the next line. This process is applied iteratively to the lines just generated and all subsequent lines to generate the next set of points. Paradoxically, this algorithm generates from the original line an infinite number of points by subtracting points from a single line, creating what is referred to as Cantor dust (Figure 1a). In principle, the algorithm can be run backwards to reform the input. The fidelity of the reconstruction depends on how well the dust is reassembled into parts and then into the whole. The scheme is robust as any missing information can be obtained from the redundant elements of each layer. The analogy here is between the dust and the synapses that form memories of an input to the brain. The process shown in Figure 1a is symmetrical. However, despite the excellent recall, the Cantor algorithm is not designed to learn. Conceptually, the scheme could be modified by using an asymmetric algorithm that removes a subset of points to generate instead a barcode specific for each input (Figure 1b). Intraconnections within each layer could ensure that each unit stores the same barcode to ensure that recall remains robust. Then the system is capable of uniquely representing each input with a barcode and is also capable of learning the relationship between input and barcode. Sharing of the same barcode across different sensory inputs helps correlate the features detected by each. Memory then represents the barcode of active synapses at each layer, and is based on the connection of synaptic units within and between layers. The wiring ensures that the memory is robust even when there are missing inputs, as the barcode in that unit can be recreated by using information from adjacent units. Loss of memory is only apparent when many of the units in a layer are no longer functional, with the gaps in output filled incorrectly, or not at all.

Modeling memory. (a) Cantor sets illustrate how information can be stored and regenerated from a set of points (corresponding to synapses) using a symmetrical algorithm to generate and reconstruct memory. (b) Here, an asymmetrical algorithm generates barcodes at each layer corresponding to the input. Syncing the barcode with neighbors ensures robust performance, even when inputs are missing. In this example, the output regenerates features absent from the original input. Sharing of the barcode across senses enhances the correlation between the features detected by each.
The simple mathematical analogy finds a biological basis in the work of Eric Kandel, whose work on Aplysia revealed two synaptic components essential for memory creation and recall.5,6 The first is the branching of dendrites to increase interactions between sensor and effector neurons, and the second is the interneurons that spread laterally to integrate inputs from other sensory pathways to enhance or inhibit the current and future effector responses. The neural network design is easily evolved in a modular fashion by adding vertical neuronal columns and lateral connections to optimize outcomes. Most often, this means operating the ensemble close to criticality, so that only certain inputs are propagated to drive a particular response. In other situations, a failure to respond or to limit a response may have negative consequences.7,8
What is the physical implementation of such a simple model of neural connectivity? Many schemes are possible. Spike trains offer one way of transmitting barcodes vertically between layers. The information is encoded by the order in which neurons in the horizontal layers fire, reflecting recent experimental findings that describe the processing of inputs by neuronal arrays. 9 The synchronization within layers is by non-spiking neurons and mostly depends on their local connectivity. The synapses involved are plastic and adaptive, with different time scales for the synaptic optimization of vertically and horizontally orientated neurons. The vertical neurons allow rapid responses, while the horizontal connections allow the efficient tuning of circuits to improve subsequent performance, as recently modeled with neural networks. 10 The system, once trained, can regenerate features absent from the initial input. However, this process becomes less reliable as more units in a horizontal layer are lost or when inputs lack sufficiently detailed information to reconstruct the stimulus.
The overall architecture of the design in Figure 1 is fixed and could be specified biologically through the interaction of the genetically encoded parts during development. There are many steps at which memory is specified in this model that could be disrupted, both genetically and by environmental inputs. For example, memory formation may be impacted by gene variants that do not generate the correct neuronal architecture, diseases that affect the interlayer conductivity of neurons as occurs in demyelinating diseases such as multiple sclerosis, disruption to the axonal transport of transcripts from the nucleus to the synapse, and outcomes that interfere with synapse formation and function.
Here, I focus on how immunological responses impact synapse formation, focusing on the ancient pathways that date back to the first unicellular eukaryote and are regulated by the unusual left-handed Z-RNA and Z-DNA nucleic acid conformations. 11 The sequences that change conformation under physiological conditions are called flipons, and are encoded by the repetitive genome that was once referred to as “junk”.12,13
Synapses and Memory
The encoding of memory through alterations in synaptic strength rather than by bioelectric fields is now widely accepted. 5 The process involves synaptic translation of mRNAs,14,15 and produces long-lived signaling complexes that strengthen synaptic connections and share similar features with prions. 16 The best characterized example is CPEB3 (cytoplasmic polyadenylation element-binding protein 3), which increases mRNA stability in the synapse by adding additional adenosines to the message tail to activate otherwise dormant transcripts. 17 The assembly of activated CPEB into a larger structure depends on a change in protein conformation that is thought to induce the same change in other CPEB monomers to grow the polymer in the same way prions extend.18,19 This active structure is spatially restricted to the activated synapse and is not transmitted to other neurons, unlike the disease-associated prions that cause neurodegeneration. Translational regulation in the postsynaptic neuron is an essential part of memory formation, as it is the enlargement or shrinkage of the postsynaptic spine that is associated with long-term potentiation (LTP) or depression (LTD) of memory. 20
Translation and Memory
Translation is also subject to regulation during immune responses. The pathways involved are triggered by the double-stranded RNAs (dsRNAs). The A-RNA double helix is usually formed by the complementary base-pairing of inverted repeats within the same transcript. The dsRNAs may originate from viral infections or from stressed, uninfected cells (Figure 2). The increased levels of dsRNA are sensed by protein kinase RNA (PKA, encoded by EIF2AK2), with the enzyme then phosphorylating the eukaryotic initiation factor 2A (EIF2A, encoded by EIF2A) to inhibit translation of RNAs that have a 5' 7-methylguanylate cap 21 (Figure 2). This outcome can inhibit memory formation.22,23 Interestingly, another set of eukaryotic initiation factors, such as EIF4G2, enables translation of some RNAs from an internal site.24 -28 The EIF4G2-dependent protein products offset the effects of cellular stress and virally encoded proteins. 27 They also enable the remodeling of synapses, 28 presumably to prevent viruses from infecting downstream neurons. Activation of PKR is also associated with Alzheimer’s disease.29,30
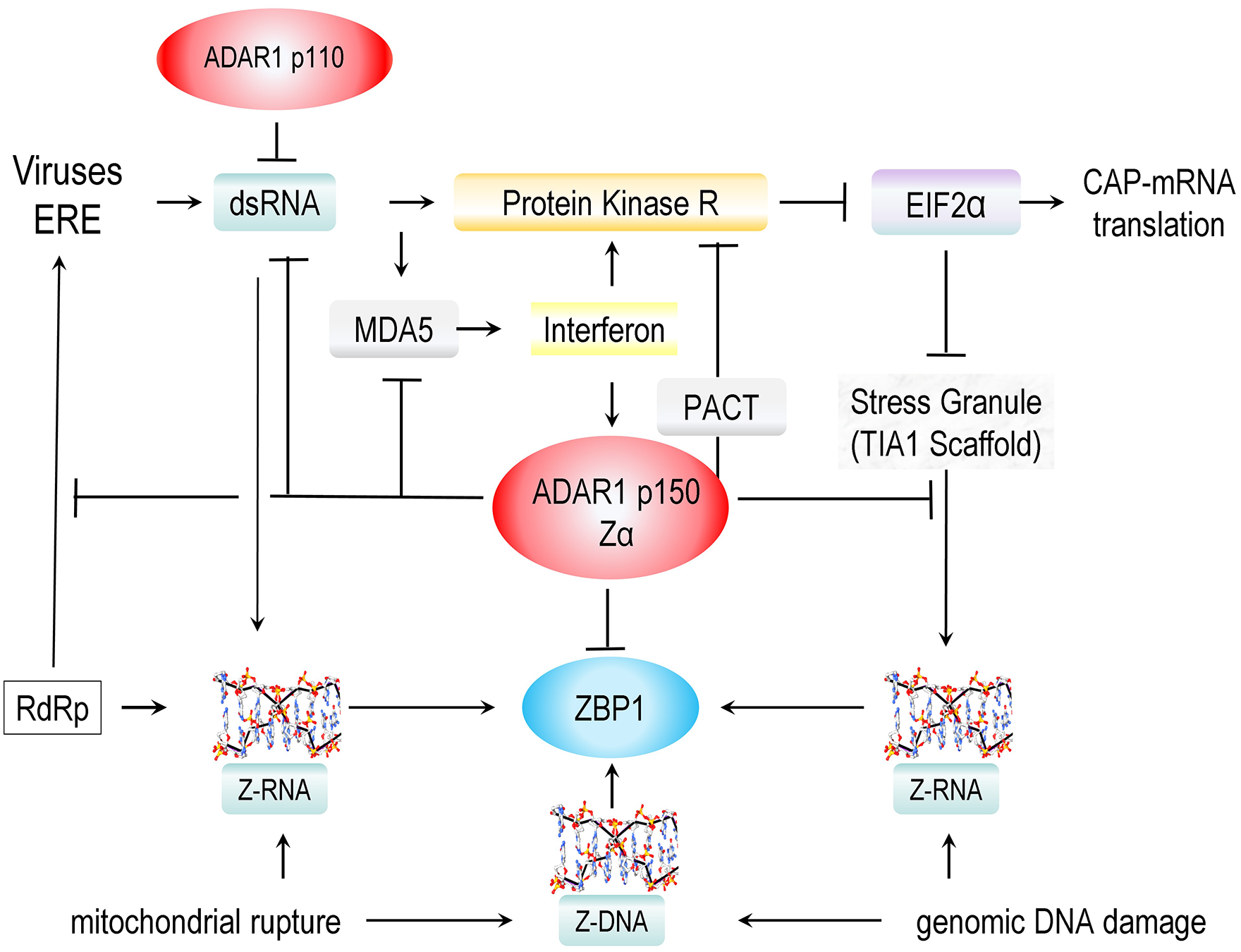
The complex pathways involved in the response to double-stranded RNAs produced by viruses and endogenous retroelements. In normal cells, dsRNA is sensed by the constitutive p110 isoform of ADAR with effects on splicing and editing of nuclear RNAs. In the cytoplasm, dsRNA is sensed by MDA5, inducing an interferon response that drives the expression of PKR, the p150 isoform of ADAR1, and ZBP1. Sensing of dsRNA by PKR leads to phosphorylation of EIF2α, inhibiting translation of capped mRNA and leading to the formation of stress granules. Cytoplasmic ADAR1 p150 is a negative regulator of these responses, binding and editing dsRNA, binding Z-RNAs, and forming complexes with helicases and proteins like PACT (Protein Activator of PKR, encoded by PRKRA) that oppose further activation of MDA5 and PKR. The binding of Z-RNAs formed in dsRNA regions and instress granules diminishes the activation of ZBP1. The proteins involved are further described in the text, and additional details are given in the following figures. ZBP1 is also activated by ZNAs released from damaged mitochondria. ZNAs also accumulate during nuclear DNA replication, especially when a cell is stressed or exposed to radiation or other mutagens.
This PKR pathway involves two other prion-like molecules: MDA5 (melanoma differentiation-associated protein 5, encoded by IFIH1) and TIA1 (T-cell intracellular antigen 1, encoded by TIA1) (Figure 2). MDA5 binds stochastically to form extended assemblies on dsRNA to initiate interferon responses that promote PKR expression, 31 while TIA-1 binds and assembles mRNA into stress granules. 32 Recent findings have connected PKR biology and the MDA5 and TIA1 prions to a novel way of encoding genetic information. The outcomes are regulated by sequences within the repetitive genomes previously characterized as “junk” DNA. The repeat elements involved are called flipons. They can adopt unusual DNA structures such as Z-DNA and G-quadruplexes inside living cells.12,13
Those flipons that adopt the left-handed Z-DNA and Z-RNA conformation (collectively known as ZNAs) play essential roles in interferon-mediated immunity. ZNAs form from right-handed B-DNA or A-RNA helices by flipping the bases over, causing the backbone to “zig-zag”. The transition does not require strand breakage or any alteration to the nucleotide sequence. 3
Z-RNAs and Immunity
Sequences that can form Z-RNA under physiological conditions classically consist of alternating purine/pyrimidine repeats, with the propensity to flip to the Z-conformation decreasing in the order CG>TG = CA > TA. The TA dinucleotide repeat is more prone to form other non-Z conformations. 34 Z-prone sequences are enriched in the Z-Box of Alu elements, a class of SINE repeat (short interspersed nuclear elements). 33 The Z-Box has a high propensity to flip to Z-RNA when Alu inverted repeat sequences (AIRs) pair to form dsRNA, as confirmed by structural studies. 35 There are over I million Alu sequences that constitute about 11% of the human genome. 36 These elements replicate by a “copy and paste” mechanism using proteins encoded by LINEs (long interspersed nuclear elements) and insert themselves into active genes, posing an existential threat to the survival of the species. 37 Many host transcripts include these sequences, usually in untranslated regions. Alus are now pressed into service to tag host transcripts as self, exploiting the fact that viruses do not contain these sequences. The former enemy now defends the genome against attack by discriminating friend from foe.
Z-RNAs are also encoded by LINEs, along with other endogenous retroelements (EREs) present in the repeat genome. These EREs lack the capacity to make the envelope proteins employed by retroviruses to bind and infect cells. The Z-RNAs formed by those EREs within host-encoded transcripts prevent the induction of immune responses against self by engaging the dsRNA-specific editing enzyme ADAR1 (encoded by ADAR).38 -43 Once bound, ADAR1 deaminates adenosines within the dsRNA regions to form inosine44,45 (Figure 3), destabilizing the dsRNA regions of some, while enabling inosine-specific nucleases to eliminate others. 46 This process prevents host dsRNA from activating MDA5 to form the prion-like filaments needed to induce an interferon-dependent response.

ADAR1 p150 and ZBP1 are the only two proteins in the human genome with a ZNA structure-specific Zα domain. Both proteins are induced by interferon. (a) ADAR1 p150 negatively regulates interferon responses by inducing the disassembly of MDA5 filaments, and by destabilizing dsRNAs through adenosine to inosine editing. (b) ADAR1 also squelches binding of ZBP1 to ZNAs. The interaction prevents the activation of ZBP1. ZBP1 induces a number of responses listed on the fight that protect the host from infections and cancers. These outcomes are detailed in Figure 6.
ADAR1 and ZBP1, and Interferon Responses
The way Z-RNAs halt MDA5 filament formation is quite elegant. MDA5 binds to RNA stochastically to nucleate filament formation. The dsRNA bound by MDA5 is overwound,47,48 rendering the dsRNA between adjacent MDA5 assemblies underwound. The underwinding is sufficient to flip right-handed A-RNA to Z-RNA. In this process, the shorter dsRNA helix of 24.6 Å is replaced by the longer Z-RNA helix that has a pitch of 45.6 Å (Figure 3a). 41 This effect can be mechanically modeled in vitro using magnetic tweezers to untwist double-stranded DNA templates.49,50 This relaxation of the tension in the dsRNA by the flip produces a conformational change in MDA5 that triggers ATP hydrolysis by the helicase. The energy released powers the flip to Z-RNA and the detachment of MDA5 from the dsRNA, 48 terminating filament formation before an interferon response is induced.
This mechanism depends on the long, interferon-induced ADAR1 p150 isoform that recognizes ZNAs through the structure-specific Zα domain.51-53 These transient, Z-RNA-dependent events that localize p150 to dsRNA can be tracked indirectly by the longer-lived inosine modification arising from the editing of dsRNA substrates. Such edits are reduced by p150 Zα loss-of-function mutations that are by now well characterized. 54 Similar loss-of-function Zα variants are causal for the Mendelian Aicardi Goutières Syndrome Type 6 (AGS6) interferonopathy and for Dyschromatosis Symmetrica Hereditaria of humans, 55 and in mouse models of AGS6.56-58 In addition to the Zα variants, other human ADAR1 variants within the catalytic deaminase domain also produce AGS6. 59
Stress granule assemblies are also associated with Z-RNA formation, which is powered by the tension generated by the hybridization of different RNAs with sequence matches. The Z-RNAs arise primarily within the Z-boxes of SINE and LINE elements, both present at high frequency in the human genome and transcribed in both forward and reverse directions.60-62 Replicating LINEs can also form Z-RNA/Z-DNA hybrids in cytoplasmic granules due to the action of reverse transcriptases, as shown by the use of small molecules that selectively inhibit this class of polymerase.41,63 The fate of stress granules depends on the context. Those formed in the absence of infection can reversibly store the RNAs to allow the rapid reinitiation of translation once the stress dissipates. However, during infection, the stress granules can activate ZBP1 (Z-DNA binding protein 1, encoded by ZBP1), the only other protein in the human genome with a Zα domain. Like ADAR1 p150, ZBP1 is induced by interferon and can induce a number of different ZNA-dependent responses when ADAR1 is overwhelmed or inactivated. These outcomes arise from regulated cell death by both necroptosis and apoptosis,64-71 through neuronal metabolic and autophagic resets,72-74 and by activation of pathways that depend on NF-κB (nuclear factor kappa B subunit 1, encoded by NFKB1). 75
Flipons and Host Survival
The strategy to control infections based on host transcripts differs from that used in other species, such as Caenorhabditis elegans and Drosophila melanogaster. In C. elegans, active retrotransposons are counterselected and represent much less than 1% of the genome.76,77 Infectious agents are eliminated by pathogen-specific guide RNAs (gRNAs). These responses require small interfering RNAs and a reverse transcriptase to amplify the gRNAs. The amplification of EREs is prevented, and their elimination promoted, by ERE-specific gRNA encoded in the genome. In D. melanogaster, EREs are more frequent and comprise ~30% of the genome. The flies use germline-encoded gRNAs amplified by either a ping-pong or a phased amplification scheme to generate piwi RNAs (piRNA) without needing a reverse transcriptase.78,79 The process depends on a DNA-dependent RNA polymerase to transcribe islands in the genome where ERE are abundant, facilitating the amplification of piRNAs, 80 or to produce dsRNAs from circular DNAs derived from pathogen genomes. 81 Because of the existential risk posed by EREs, other metazoa also avoid retention of reverse transcriptases that would further promote the spread of retrotransposons. These enzymes were long ago selected against in humans. 1 Instead, the host-based defense based on Z-boxes within EREs provides a mechanism to protect against a wide range of known and emerging threats and cellular dysfunctions. 11 In response, human neurotropic viruses, such as Herpes Simplex, have evolved strategies to circumvent such interferon-dependent responses.82,83
Like ADAR1 and ZBP1, host transcripts that form ZNAs are also induced by interferon during viral infection. In addition, other processes give rise to ZNAs. Both Z-DNA and Z-RNA are formed in stressed or cancerous cells and released from damaged mitochondria,84-87 or arise from subtelomeric regions during a replicative crisis. 88 These ZNA-forming transcripts are often derived from repeat elements like those produced during infections. These different flipon sequences play an active role in defending the cell and are subject to selection during evolution. Those flipons that protect and enhance the reproduction of their hosts are transmitted to the next generation. 89
Roles for ADAR1 in Normal Brain Function
While ADAR1 helps protect the host, new uses for this enzyme have evolved. The A-to-I RNA modification plays a beneficial role in learning through the recoding of codons. These outcomes involve the RNA editing of neurological receptors. In healthy brains, the glutamate ionotropic receptor AMPA type subunit 2 receptor (encoded by GRIA2) undergoes recoding at the Q/R site. 90 Over 99% of transcripts from this gene are edited in adults. Loss of editing alters the calcium permeability of the receptor and leads to dendritic spine loss, neurodegeneration, and impaired memory.91,92 Deficient editing is also associated with amyotrophic lateral sclerosis (Kwak, 2005 #3658). Mouse studies have also linked A-to-I mRNA editing of the serotonin 5-hydroxytryptamine receptor 2C (encoded by 5-HT2CR) to decreased anxiety- and depression-related behaviors. 93 Editing is associated with conformational changes in the receptor that decrease signaling. Animals resistant to a traumatic event showed higher editing in the central amygdala than control animals. 94
ADAR1 can also affect memory by editing non-protein-coding transcripts (ncRNAs). In particular, the interaction of ADAR1 with EREs and the docking of ADAR1 p150 to Z-DNA played a key role during fear extinction learning. 95 The sites bound were associated with repeat elements. ADAR1 knockdown impaired memory updating. During fear conditioning, Z-DNA formation was enhanced at active genes whose transcription was triggered by the training. In contrast, extinction-trained mice had reduced Z-DNA formation and showed increased ADAR1 editing. Mutation of the Z-DNA binding domain or a catalytically inactive enzyme prevented the updating of fear responses. An analysis of transcripts during extinction revealed an overrepresentation of SINE/LINE elements at DNA sites engaged by ADAR1. The results suggest that editing EREs is essential for rewiring memory circuits. Without the p150 ADAR1 isoform, conditioned fear responses persist. These processes occur in neurons that express the activity regulated cytoskeleton associated protein, encoded by the ARC gene.
Memory EREs as Ribotransmitters
The deletion of the ARC protein impairs neural plasticity in mice. It is derived from the LINE and retroviral classes of retrotransposons that encase their RNAs in protein capsids encoded by group antigen genes (GAGs). Interestingly, the genome contains many different GAG remnants. These capsids have been exapted to serve important functions in animal biology. In certain clades, GAGs encode the syncytin protein essential for placental formation.96,97 The ARC protein is another example. The ARC capsid can encapsulate its own RNAs and transport the transcript across a synapse. The capsid can incorporate RNAs for reporter proteins as it lacks the domain that gives specificity by restricting the interaction to its own RNA. Indeed,98,99 Other genome-encoded GAGs can also incorporate RNAs into capsids. These RNAs’ neurological expression is depleted upon deletion of the ERE gene. Currently, the function of these RNAs is poorly characterized. 100 The capsids could transport RNAs across the synapse and modulate memory formation after translation in the post-synaptic spines. 11 Consequently, the effects of these ribotransmitters on the downstream neuron are rapid and localized to the recipient spine. The ribotransmitters then coordinate the activation state of both neurons to either strengthen or weaken the synapse. The presynaptic neuron has a dual role. It sends transcripts across the synapse that instruct the postsynaptic neuron on what to make, and it encodes the receptors that detect the response generated. This pathway enables the upstream neurons to program the neurons it connects with. 101 This design exploits the extreme plasticity of hippocampal granule cells and allows the generation of different connection patterns. 102 The connection made depends on how the postsynaptic neuron responds. From an evolutionary perspective, natural selection of the ribotransmitter-dependent feed-forward mechanism depends on the transcripts expressed by the upstream neuron, and whether the programs enacted improve the probability of propagating progeny.
Memory versus Immune EREs
Retrotransposons and viruses can also jump the synapse, potentially using a similar mechanism to that proposed for ribotransmitters. The transmission of immune EREs from the pre- to post-synaptic neuron can protect against the dissemination of these agents by inhibiting translation of their messages in the postsynaptic spine. The response is again rapid, allowing time for the post-synaptic neuron to respond in a more robust manner by expressing interferon-stimulated genes, including EREs. Indeed, the enhanced expression of Alu elements has been noted in neurological disease.29,103-105 The induction of interferon-induced genes and transcripts during viral infection overwhelms the ability of ADAR1 in the presynaptic neuron to negatively regulate the MDA5-dependent interferon responses (Figure 3) and also prevent the PKR-mediated shutdown of translation 39 (Figure 4). In these cases, ADAR1 could also fail to limit transmission across the synapse of immune ERE that protects a post-synaptic neuron from infection. Whether such transmission of immune EREs between neurons occurs requires experimental investigation.

Ribotransmitters and Memory formation. In this model, memory and immune ERE (endogenous retroelement) RNAs regulate translation of transcripts in boutons and spines by modulation of protein kinase RNA (PKR) activity. PKR otherwise switches the eukaryotic initiation factor usage from EIF2α to EIF4G2, leading to the preferential translation of stress-responsive mRNAs. Memory EREs enhance synaptic connectivity while immune ERE dismantle connections to prevent transmission of retrotransposons and viruses across the synapse. ADAR p150 binds Z-RNAs to negatively regulate interferon response by inhibiting MDA5 filament formation and editing immune ERE. PKR also mediates the phosphorylation of Tau, altering the release and uptake of synaptic vesicles. Downregulation of these processes would limit the spread of viruses. Encapsulating pathogens and viral factories in β-amyloid (Aβ) cages would disrupt their replication and enhance intracellular immunity.
Interestingly, the activation of PKR by dsRNA affects memory through an additional pathway: the phosphorylation of Tau (encoded by MAPT, microtubule-associated protein tau). Tau plays a role in synaptic vesicle recycling.106,107 Inhibition of Tau by phosphorylation of threonine 181 (ptau118) by PKR (and other kinases) 108 likely inhibits the release and uptake of viral particles by neurons, thus preventing the spread of pathogens via retrograde transport. The accumulation of intracellular β-amyloid is also seen in mouse models of AD. 109 The encapsulation of pathogens and viral factories in amyloid cages would further disrupt their propagation (Figure 5). Notably, an association of Herpes simplex virus type 1 DNA with amyloid plaques in AD patient brains is reported. 110 Varicella Zoster also elevates intracellular levels of β-amyloid. 111 The uptake and clearance of other protein aggregates will also be impaired when the recycling of exosomal vesicles is impaired. Interestingly, phosphorylated tau accumulates in AD, tauopathies, and other neurodegenerative diseases.108,112-114 Indeed, the accumulation of amyloid beta (Aβ) across multiple brain regions in AD is associated with plasma levels of tau threonine 181 (ptau118), a site phosphorylated by PKR, and other kinases. 115

The structure of Aβ1-42 from Protein Database Entry 2NAO. (a) Here, the structures are positioned to emphasize the β-sheet stacks and their potential to assemble into extended arrays. (b) The positions of Aβ1-42 residues. (c) The potential contacts between dimers of Aβ1-42 are involved in forming arrays. The sequence below highlights the E22 and K28 residues involved in the contacts. (d) The location of the Aβ42 peptide in the Amyloid-beta-precursor protein. The wild-type sequence of the peptide is shown along with a set of sequence variants that affect the risk of familial Alzheimer’s Disease. The variants are located where the amyloid monomers contact each other. The position of E28 is highlighte in red and K28 in blue.
Overall, such ERE-dependent responses against infectious agents come at the cost of impaired memory. Halting the retrograde spread of infectious agents by disassembling synaptic connections between neurons would ultimately lead to cognitive decline during chronic infections. The production of immune ERE during stress responses from various origins, such as telomeric or mitochondrial damage, while not causal in these cases, may nonetheless contribute to the pathology by releasing interferons and initiating inflammatory cell death.71,84,87,88
Memory, Complement, and Amyloids
Memory loss and impaired cognitive status are further exacerbated by the activation of microglial phagocytosis by ERE and infection.72,116 The responding microglia prune dendrites coated with iC3b, the proteolytic product of complement C3. The deposition of iC3b is triggered by complement C1q (encoded by C1QA, C1QB, and C1QC) activation.117-119 Other non-infectious, oxidative, stress-induced, and immune-related pathways also cause loss of synaptic contacts and neuronal loss through complement and microglia activation.120,121
A number of infectious agents have been proposed as causal for cognitive decline.122-126 Indeed, a brain bacterial microflora has been proposed as an essential contributor to disease, although the association is still considered tentative.127,128 The recently discovered neural glial lymphatic system likely protects the brain from bacterial infections.129-131 In immunocompromised hosts, resident bacteria may exacerbate memory loss through synaptic pruning due to the persistent activation of complement. Here, extracellular flipons play an unexpected role. The biofilms produced by bacteria contain both Z-DNA 132 and G-quadruplexes. 133 These nuclease-resistant structures act as an exoskeleton to protect the bacteria against host defenses. 24 The biofilms induce antibodies that contribute to neurological dysfunction through complement activation. Clinically, some outcomes are seen in autoimmune diseases, such as systemic lupus erythematosus. 134 The bacterial amyloids also produce inflammation and microglial activation by engaging toll-like receptors. 135 The response promotes clearance by phagocytosis of both the bacteria and the amyloids.
Bacterial and Host Amyloids
The bacterial amyloids may seed the aggregation of other proteins prone to prion formation, such as synuclein in Parkinson’s disease. 125 Most often, the bacteria implicated are inhabitants of the gut that enter the circulation through a leaky gut wall.124,125 Normally, the entrapment of these bacteria is facilitated by neutrophils and the extracellular nets they form by extruding their genomic DNA. The oxidative bursts generated by neutrophils then kill the bacteria. The process also fragments the Z-DNA that holds the biofilm together, enabling clearance of the bacterial bunker by the immune system. 133 However, the ability of neutrophils to enter the brain and form these protective extracellular nets is limited by the high selectivity of the blood-brain barrier (reviewed in Santos-Lima et al 136 ). The exclusion of neutrophils protects the brain from the oxidative damage they cause. Instead, others have proposed that the human amyloid beta protein precursor (encoded by APP) entrap bacteria in the brain. The cages formed are equivalent to neutrophil nets used in other tissues and seed the formation of amyloid plaques.122,123,126 The plaques incorporate the proteolytic Aβ fragments of APP. These Aβ seeds may also arise in other tissues and constrain the growth of bacterial biofilms, most notably in the mouth and gut. The seeds may also be transported to the brain via the blood. 137 Normally, the neural glial lymphatic system filters them out. When this clearance system fails, the risk of plaque formation in the brain increases.
The Genetics of Host Amyloids
The question arises of how such an infectious origin of plaques connects with the well-known genetics of AD. A number of Aβ sequence variants are associated with familial forms of Alzheimer’s disease. The amino acid changes can alter the folding of Aβ-derived peptides (Figure 5). 138 The structure of Aβ42 has been characterized139,140 and solved in the presence of zinc ions. 140 The peptide folds to form a β-sheet stack composed of Aβ dimers (Figure 5a, as labeled in 5b). Interestingly, these dimers have the potential to assemble orthogonally into a sheet of various types,141,142 although the particular structure shown in Figure 5c has not yet been experimentally visualized. This assembly aligns E22 (where E is the single-letter code for glutamate) with K28 (lysine 28). Notably, E22 variants are found in many of the early-onset AD families. In the context of the bacterial hypothesis, these variants may induce amyloid aggregates that are prone to breaking, creating fragments that enter the circulation and seed amyloid formation in the brain. The fragility of amyloid assemblies enables bacteria to evade elimination, leading to chronic infections and the exacerbation of plaque formation.
Flipons and the Introspective Cell
Not unexpectedly, the etiology of infection and inflammation of memory decline in neurological disease is complex, with many pathways contributing to the detrimental outcomes. New insights are provided by recent discoveries showing that cells monitor ZNA levels to assess their state of health and initiate corrective actions. There are many reasons why these findings were previously missed. The most obvious one is the experimental challenge of working with a dynamic and energetically unstable conformation. It took some time to solve these problems. 143 Another difficulty is that the “classical” genetic models in which diseases are traditionally modeled, such as the roundworm Caenorhabditis elegans and the fly Drosophila melanogaster, implement immunity through a different strategy: they target infectious agents using pathogen-specific gRNAs rather than with host-encoded RNA proxies. Those model organisms rely on reverse transcriptases that are selected against in animals, trying to thwart the attack by invasive retroelements enabled by this class of enzymes. 11
A further challenge is that ZNA-related phenotypes are most evident under stress conditions, especially those that persist over time. The scaffolds in normal neurons that mediate responses (Figure 6a) are distinct from those in stressed cells (Figure 6b). While the components are genetically encoded, the environmental events that trigger their assembly are not (Figure 6). Overall, the scaffolds in each state differ in their construction and the outcomes produced. The responses depend on the non-templated conjugation of ubiquitin-like proteins to other proteins, the localization of specific complexes to two-dimensional membrane surfaces, and the formation of large protein assemblies from simple amino acid motifs. 74 In normal cells, these scaffolds help match cellular state with metabolic needs. They maintain the structural integrity of cellular membranes while enabling their remodeling. Different scaffolds form when cells are stressed, altering the transcriptional and translational responses to external perturbations. The newly formed structures also change how cells communicate with each other.

ZNA-dependent phenotypes in stressed cells. (a) Different non-genetically templated scaffolds are used in normal cells, compared to (b) those scaffolds present in stressed cells, as described in the text. (c) The inverted U-shaped dose-response curve, where functional outcomes vary with a therapeutic’s overall mechanism of action. Interferon and complement limit infection at lower doses; they disrupt memory at higher levels. Loss of amyloid protects against plague formation but not against bacterial infection. At high levels, cellular stress is neurotoxic. The balance between protection and loss of neural function likely varies by brain region and by individual.
The level of EREs present enables a cell to monitor its well-being and respond accordingly. Higher ERE expression leads to increased dsRNA formation and the persistent formation of Z-RNAs within a cell (Figure 3). Unchecked, an increase in ERE levels due to infection and stress activates innate immune responses through Z-RNA-induced cell death pathways. Lower levels of Z-RNA instead support the proliferation and differentiation of cells (Figure 6). The tissue-specific deletion of ADAR p150 can disrupt these pathways. The higher Z-RNA levels that result impair the differentiation of CD4+ helper cells within the thymus, 144 the formation of germinal centers, 145 the maintenance of pancreatic homeostasis, 146 normal cardiac development, 147 and maturation of neural crest-derived melanocytes and Schwann cells. 148 Disease outcomes reflect the scaffolds formed in each tissue. Neural aplasia may result when altered turnover leads to protein insufficiency. The accumulation of toxic aggregates stems from the failure to clear cellular complexes. Senescent progeny may reflect a failure to execute dysfunctional cells, leading to the accumulation and crowding out of younger cells. Paradoxically, these responses lower the risk of cancers in AD.149,150 While multipotent neural stem cells do enable the replacement of damaged neurons in AD, 151 their extreme plasticity increases the risk of devastating tumors like glioblastomas. 152
Therapeutic Approaches
Overall, the repetitive genome contributes to the complexity of memory loss in neurodegenerative diseases, with flipons playing many different roles. Z-RNAs encoded by ERE shape memory formation by modulating cellular responses to stress and infection. Forming alternative nucleic acid conformations such as Z-DNA, Z-RNA, and G-quadruplexes limits the spread of bacteria, viruses, and retrotransposons from one neuron to the next and from one brain area to another. There is a cost as the synaptic connections break down and neurons shut down translation of the proteins needed to form new associations. At worst, the defensive responses turn memories into Cantor dust with a chaotic collapse of cortical connectivity. This outcome becomes progressively more likely as the reconstruction of responses by the surviving interconnected neuronal modules fails to compensate for the loss of interconnecting neurons. Instead of a robust, redundant, and distributed information storage, the brain attempts to make sense of inputs from what remains connected. Moderation of affective responses will also be impaired by the loss of neurons that tune those circuits.
Like other successful health measures, developing vaccines that produce neutralizing antibodies to thwart neurotrophic viruses is one strategy to prevent the infectious cycles that predispose to memory loss. Another prevention measure is the appropriate treatment of chronic bacterial infections likely to spread blood-borne amyloid seeds to the brain. Measures to counter excess complement activation in the brain may ameliorate progression. 153 The use of non-complement activating antibodies to prevent the spread of amyloid seeds to the brain is possible, provided sufficient levels are maintained. Overall, the development of therapeutics is challenging due to the inverted U-shaped dose-response curves. For example, lowering amyloid may increase the production of complement-fixing antibodies against pathogens. Optimal dosing will likely vary by individual and the underlying causal factors (Figure 6c). The question of whether ribotransmitters that are transported across synapses by exapted retrotransposon capsids play an important role in neuroplasticity and neural regeneration is currently unanswered. If they do, developing therapeutic approaches to enhance their production or employing these transporters to deliver RNA messages to the targeted neurons offers a promising area to explore. The idea of promoting cognitive recovery rather than treating disease is fast becoming a new paradigm in the field. 154
Footnotes
Author Contributions
This work was conceived, written, and illustrated by the author
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Generative AI
The author declares that no generative AI tools were used in the production of this manuscript
Data Availability Statement
All data and methods are available or described in the main text or the references.