
Abstract
Here, we describe two congenitally deaf male siblings with the same compound heterozygotic, likely pathogenic mutations in the FGF3 gene, associated with the labyrinthine aplasia, microtia and microdontia (LAMM) syndrome. Both children had bilateral cochleovestibular aplasia, precluding cochlear implantation. The elder brother received an auditory brainstem implant (ABI) with very limited auditory responses. During the ABI-surgery of the younger subject, it was discovered that excellent auditory responses could be obtained when the electrode array was placed considerably more caudally and more medially than standard. It was observed that the foramen of Luschka, the entrance to the lateral recess of the fourth ventricle was located more caudally. In view of this observation the good auditory development of the latter child, it was decided to give the older child a contralateral ABI. Again, it turned out that the anatomy of the brainstem was abnormal with a more caudal location of the foramen of Luschka and the cochlear nucleus, and this child is showing good progress with his auditory development. It is concluded that one should be aware of the anatomical differences at the level of the brainstem when placing an ABI in children with this genetic disorder (and most likely also in the LAMM syndrome). This also underpins the need of a multidisciplinary approach with closely collaborating team members and good family guidance when diagnosing and treating children with rare deafness.
Plain language summary
This case series describes three placements of Auditory Brainstem Implants (ABIs) in two congenitally deaf brothers with a gene defect associated with the Labyrinthine Aplasia, Microtia and Microdontia (LAMM) syndrome. In line with this syndrome, they both had no inner ears and no hearing nerves and therefore couldn’t be helped with a cochlear implant. The only option was an ABI, with a mesh electrode positioned on the cochlear nucleus, the first relay station of the hearing system in the brainstem. The first ABI electrode in the elder brother was placed in the classical position and did not yield much benefit. During the ABI surgery in the younger sibling, it was discovered that the location of the cochlear nucleus (the target site for an ABI) was located further down and the electrode had to be inserted deeper. This child showed a good auditory development, and it was decided to give the older child an ABI at the other side. Again, it turned out that the anatomy of the brainstem was abnormal with a lower location of the cochlear nucleus. With this second implant, this child’s auditory development is showing good progress. It is concluded that one should be aware of the anatomical differences at the level of the brainstem when placing an ABI in children with this genetic disorder (and most likely also in the LAMM syndrome).
Keywords
Introduction
The FGF3 gene, 1 located on chromosome 11, encodes for fibroblast growth factor 3. During antenatal development, this protein triggers the processes leading to the development of the inner ears (cochleae). Mutations in both alleles of the FGF3 gene are therefore known to cause profound deafness due to severe forms of cochleovestibular dysplasia, including labyrinthine aplasia or Michel’s deformity. 2 Apart from this, FGF3 deficits can lead to concomitant malformations of the outer ear and small and widely spaced teeth, and then it is called the labyrinthine aplasia, microtia and microdontia (LAMM) syndrome.3–8 This is an extremely rare autosomal recessive disorder, for which no prevalence is known to date, and for which a recent literature review reported 24 pathogenic and likely pathogenic variants of the FGF3 gene. 3
The complete absence of inner ear structures in labyrinthine aplasia precludes rehabilitation with cochlear implants (CIs), devices that directly stimulate the auditory nerve with an electrode array, placed in the scala tympani of the cochlea. Therefore, the only available option to provide sound perception, and possibly the development of oral communication, is by using an auditory brainstem implant (ABI). 9 This device technically resembles a CI, but the electrode contacts are on a mesh, placed on the cochlear nucleus, the first relay station of the auditory pathway in the brainstem.
This article describes the rehabilitation with ABIs of two brothers (indicated as Child A and B) with compound heterozygotic mutations in the FGF3 gene in our tertiary referral centre. Apart from bilateral labyrinthine aplasia, they both had bilateral aplasia of the auditory nerve, making these children very suitable candidates for rehabilitation with ABIs. To our knowledge, there is very limited literature on the use of ABIs in cases with FGF3 mutations, including the LAMM syndrome. We are only aware of one case, included in a recent publication on ABI results in 38 children and a review of the literature. 10 The focus will be on the consequences for the electrode placement of the anatomical abnormalities found in the brainstem in both siblings, which may need special consideration when performing ABI-surgery in this patient population.
Auditory brainstem implants
An ABI is a specialized medical device designed to restore a sense of sound to individuals who are profoundly deaf due to non-functional or absent cochlear nerves, conditions that preclude the use of conventional hearing aids or CIs. Unlike CIs, which stimulate the cochlea in the inner ear directly to convey sound signals to the brain via the cochlear nerve, ABIs bypass the cochlea and the cochlear nerve entirely. They work by directly stimulating the cochlear nucleus (CN) in the brainstem, the part of the auditory pathway responsible for processing auditory information received from the cochlea. The CN is located in the floor of the lateral recess of the fourth ventricle (Figure 1), which is surgically accessible via a neurosurgical route, the so-called ‘retro-sigmoidal’ approach. In our centre, the procedure is performed by an ENT-surgeon and a neurosurgeon who both have ample experience with this procedure, guided by electrophysiological measurements (electrically evoked auditory brainstem responses, eABRs) by two clinical physicists who have both ample experience with such recordings. All implantations described in this study were performed with a MED-EL Mi1200 SYNCHRONY PIN ABI implant. The intraoperative measurements were performed with a Natus Nicolet EDX (Natus Medical, Orlando, FL, USA) recording device. For the measurements, aimed to find the right position on the brainstem, the so-called ‘ABI Placing Electrode’ from MED-EL was used. The placing electrode has two advantages compared to the electrode array of the ABI implant. First, it has a smooth surface, without the Dacron mesh used for stabilization of the final electrode array, reducing the chance of damaging the brainstem when it is moved to find the right position. Second, the electronics are located outside the patient, reducing stimulus artefacts.

(a) Top view of the brainstem at the level of the fourth ventricle. The dashed arrow indicates the route of insertion of an ABI via the foramen of Luschka into its lateral recess, guided by the course of the glossopharyngeal nerve (IX). In blue, the cochlea, auditory nerve (VIII) and the CN are indicated. In the cases described in this article, both the cochleae and auditory nerves were absent, creating the indication for an ABI. (b) Detail of the foramen of Luschka, the route of access to the CN, which is usually located in the extension of the IXth cranial nerve, just medial (left in the figure) to Sollman’s vein.
Sennaroglu et al. 12 were among the first to demonstrate the potential role of ABIs in providing auditory rehabilitation to prelingually deaf children with inner ear malformations, including severe stenosis of the cochlear aperture and cochlear nerve aplasia, as occurring in individuals with FGF3 gene defects (and the LAMM syndrome). Additionally, Monsanto et al. 13 offered insights into the outcomes of ABIs in children based on a review of the literature, providing further understanding of the efficacy and challenges associated with ABI use in populations. 13 The oral-aural speech and language development with ABIs in congenitally deaf children requires a much more intensive and longer lasting rehabilitation process than with CIs. The procedures used in our centre, and the differences between both programmes are the same as the ones described in detail by Van der Straaten et al., 14 who also made a comparison between the results with CI and ABI in our centre, especially in congenitally deaf children with additional handicaps.
Paediatric rehabilitation programme for ABIs
Children are considered for an ABI when imaging reveals that the auditory nerve and/or cochlea are poorly developed, or in cases of congenital malformation or aplasia of the cochlea, cochlear trauma, or cochlear ossification following meningitis. The Leiden University Medical Centre is the sole hospital in the Netherlands performing ABI surgeries for children. The ABI rehabilitation programme at our clinic is an intensified extension of our CI rehabilitation programme and consists of so-called ‘intensive weeks’ with fixed components. However, there is a significant difference in duration and intensity of the programme for CI and ABI recipients.
A general basis for the rehabilitation for children with an ABI consists out of 12 rehabilitation weeks spread over the first 2 years post-implantation, compared to 6 weeks for CI children, distributed over the first year after implantation. Research and clinical experience suggest that children with an ABI require more and longer rehabilitation due to the complexity of their condition. An intensive week includes three to four mornings of appointments with a speech therapist, audiologist and psychologist. The goal of this specialized treatment programme is to adjust the ABI and integrate its use into the child’s world and his surroundings as effectively as possible. Each intensive week follows a structured approach, including fittings (ABI tuning), auditory stimulation (focusing on expanding various phases of auditory development, encouraging vocalization through play and sometimes spoken language development), Pedagogical Interaction Guidance (using play materials to work on communicative prerequisites like eye contact, turn-taking, concentration or other areas of focus) and auditory testing with the ABI. The exact structure and intensity is tuned to the specific child, its development and the support system. We advise the parents to use sign-supported language as the preferred mode of communication with their child, as it will usually take many years for the ABI to yield sufficient oral-aural communication for use in daily life. In addition, all parents and children get education in sign language via the home support service (or schools for the deaf).
Early language development is monitored with validated questionnaires, including the IT-MAIS-NL, 15 a validated Dutch translation of the Infant-Toddler Meaningful Auditory Integration Scale, 16 resulting in a score between zero and 40, and the Categories of Auditory Performance (CAP), 17 a scale on which children’s developing auditory abilities can be rated in the following eight categories in order of increasing difficulty, ranging from (CAP = 0) ‘displays no awareness of environmental sounds’ to (CAP = 7) ‘can use the telephone with a familiar talker’.
Presentation of cases
Case A: Management and rehabilitation
Patient A was a neonate, referred to a tertiary audiological/CI centre for further evaluation following a negative outcome from the neonatal hearing screening, with the initial consideration for rehabilitation with a CI. In addition, he had bilateral ear pits, a right-sided cup ear, a mild dolichocephaly (elongated head shape), a pre-axial oligodactyly of the left thumb and was slightly hypotonic. He had no additional needs (no cognitive impairment, normal vision). Subsequent radiological assessments, including computed tomography (CT), revealed the absence of both cochleae, indicating complete labyrinthine aplasia and the bilateral absence of the cochlear nerve (Figure 2). Concurrent with these findings, genetic testing was undertaken to provide additional insights into the patient’s condition. It turned out that he had the following compound heterozygotic, likely pathogenic mutations in the FGF3 gene:
- NM 005247.2(FGF3):c.283C>T, p.(Arg95Trp), maternally
- NM 005247.2(FGF3):c.45deIC, p.(Trp16fs), paternally
From the moment, it was confirmed that the child was deaf, his parents began learning Dutch Sign Language (NGT) at home, making NGT and Sign Supported Dutch (NmG) their primary means of communication. The mother is employed as a childcare worker, and the father works as a service technician.
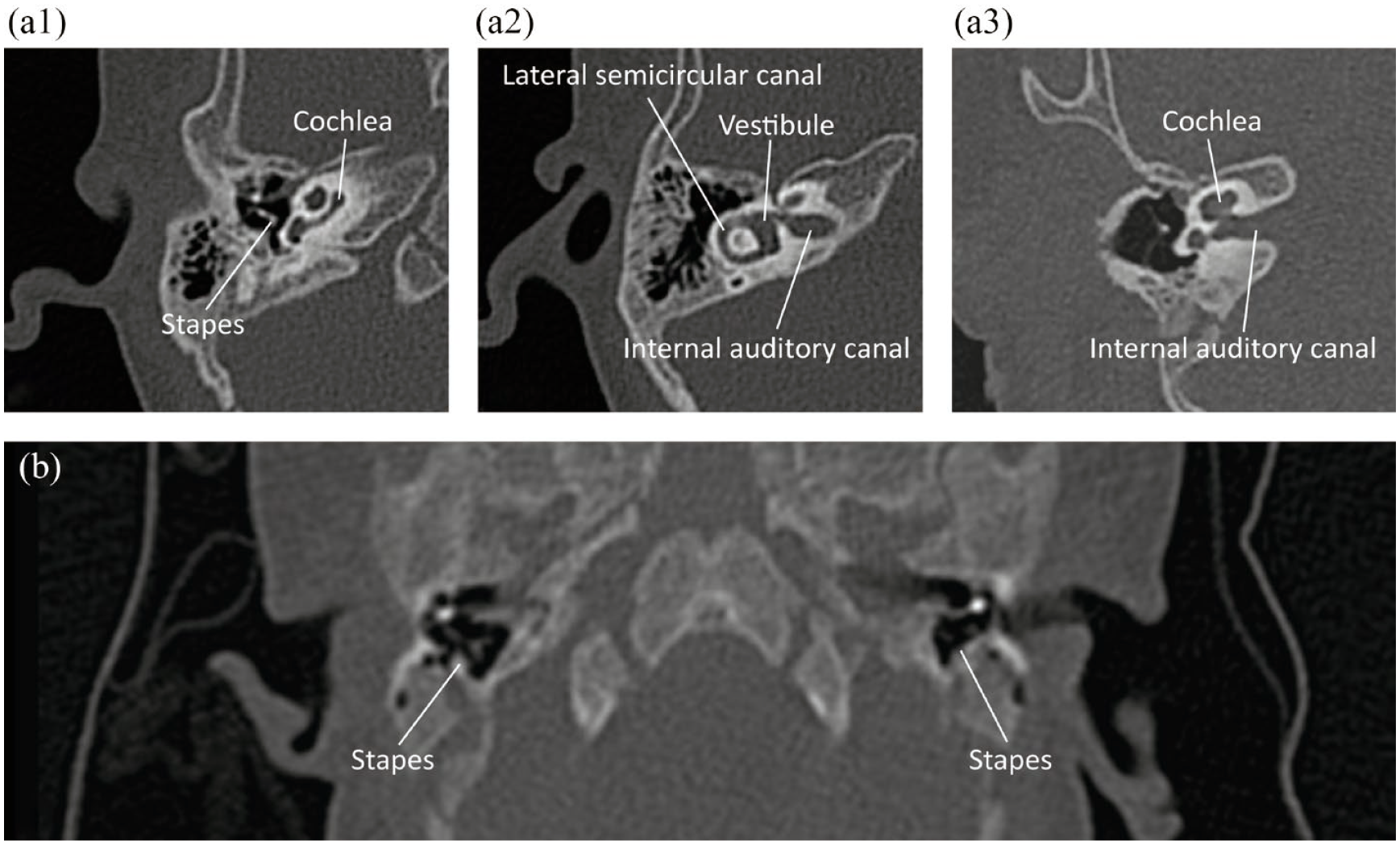
(a1) Axial CT scan of right ear of normal 1-year old child at the level of the cochlea. (a2) Axial CT scan of the same ear as in A1, now at the level of lateral semicircular canal and vestibule. (a3) Reoriented CT scan of the same ear as in A1 and A2, now at the level of the internal auditory canal. (b) Axial CT scan of child A, demonstrating the bilateral absence of the cochlea, semicircular canals and internal auditory canal, which is also not visible in other slices.
Given the absence of both cochleae and cochlear nerve, the patient was referred to our expert centre for rare ear diseases for an ABI evaluation at 4 months of age. The ABI surgery was performed on the left side when the patient was 14 months old. During the surgical procedure, the posterior fossa was small, especially in the basal direction and the complete absence of the vestibulocochlear nerve (n. VIII) was confirmed. The position of the CN was not easily found, and the placing electrode was tested in four different positions. At high current levels, one electrode pair elicited clear responses with latencies indicative of auditory stimulation. However, in several locations, a later response (4–6 ms), not associated with auditory stimulation, was observed. Based on these bipolar recordings, it was concluded that the CN was small in this case and the final electrode position was determined in such a way that the CN was expected to be in the centre of the array. With the implant set in the monopolar stimulation mode (which is the way the implant is intended to be used and which requires an irreversible action during surgery in this implant), three contacts elicited clear auditory responses, with a possible fourth showing similar outcomes. However, the stimulation levels needed to elicit these responses were much higher than usual. Other contacts elicited either no responses or responses with longer latency that was non-auditory in nature.
Throughout the rehabilitation process, it became necessary to deactivate most of the contacts due to the nature of the responses observed. Ultimately, only three contacts remained active, with stimulation provided at maximum tolerable levels. Free field testing revealed that the patient exhibited very minor reactions to acoustic stimuli, suggesting a limited but discernible capacity for auditory perception through the ABI. Hearing development remained low as shown in Figure 3 where he IT-MAIS-NLs score remained at around 3 points of a 40-point scale and CAP-score remained 0 indicating that there was no awareness of environmental sounds. Nevertheless, the child continued to wear his speech processor on a daily basis and his mother noticed that he produced more variable utterances when the implant was active.

Hearing development over time scores of both siblings, with the IT-MAIS, which has a maximum score of 40. Data of both children are plotted against the time from hookup of their individual devices, in months. Surgery of child A was at age 14 months, the implantation of child B was at the age of 13 months, the second implantation of child A was at age 61 months.
Case B: Implantation and rehabilitation of the younger brother
Child B’s hearing journey paralleled that of his older sibling, child A, only with foreknowledge of the condition. He was born when child A was 2 years and 7 months old. Following referral from neonatal screening, directly to our centre, the absence of responses on auditory brainstem response recordings and subsequent genetic testing revealed a shared genetic hearing loss with his brother. He had a right-sided limited microtia with an ear-pit and a pointed pinna of the left ear. By the age of 4 months, imaging studies confirmed child B also had complete labyrinthine aplasia and a bilateral absence of the cochlear nerve. Like his older brother, he had no additional needs.
Despite the modest outcomes observed in Child A, the parents decided to proceed with an ABI for Child B, driven by hope and the desire to explore all possibilities for auditory improvement. At the age of 13 months, Child B underwent surgery for ABI placement on the left side. Again, the complete absence of the VIIIth cranial nerve was confirmed during surgery. In this case, it became clear that the choroid plexus (see Figure 1) was abnormally located, caudally to the IXth nerve, but still marking the entrance to the lateral recess of the fourth ventricle. As a result, the placing electrode was positioned in a non-standard location during the procedure. The initial placement, which was relatively deep into the lateral recess, yielded very positive results, with clear auditory responses detected across all bipolar combinations. The success at this unusual location led to its selection for the final electrode placement, where seven electrodes demonstrated clear auditory responses at relatively low stimulation levels. The other contacts elicited responses at higher levels.
The progress at rehabilitation for Child B has been promising, with regular adjustments made to increase levels and optimize auditory input. Some electrode produced non-auditory stimulations such as arm movement, head movement and disbalance, leading to their deactivation. The remaining seven active contacts provided auditory percepts, which was further objectified during free field-testing. Child B has shown good responses to musical instruments, though warble tones gained less of his attention. Unfortunately, he refuses to wear his speech processor all day and is not too interested in sound. Auditory thresholds have been identified at around 40 dB HL, marking a significant milestone in Child B’s auditory rehabilitation. In Figure 3 the IT-MAIS-NL data of Child B are plotted, showing an increase up to a score of 11 on a 40-point scale after 2 years of ABI use. At that time, his CAP score was 1, that is, awareness of environmental sounds. He clearly outperformed his older brother, and this initiated a new view of case A.
Case A: Implantation of the contralateral side
Considering child B’s successful outcomes and the less-than-expected performance of child A, the medical team revisited child A’s case for further evaluation. The intra-operative evoked auditory brainstem responses (eABR) recordings, the fitting outcomes, the initial auditory responses and rehabilitation outcomes for child A all had not met the anticipated objectives. Child B’s surgery had led to the hypothesis that the brainstem anatomy in this specific genetic disorder was abnormal, and that it might well be that in child A, the electrode was placed latero-cranially to the CN. Inspired by the positive findings in Child B’s case and after a shared decision-making process with the parents, we decided to explore child A’s contralateral (right) ear, to see whether an alternative implant location would yield better results for the older sibling.
Child A underwent the procedure to implant the contralateral ear at the age of 5 years and 2 months. The surgical team employed a strategy similar to the one used for Child B. The IXth cranial nerve turned out to be relatively large, and the foramen of Luschka was located extremely caudally, even more than in his sibling. The placing electrode was placed in three different locations, ultimately demonstrating good to acceptable electrically eABRs at standard stimulus levels, with minimal late, non-auditory neural responses. With the final electrode placement, auditory responses were successfully elicited in monopolar mode from all contacts, although two contacts necessitated higher current levels. Notably, five contacts exhibited some non-auditory components in the neural response at elevated levels.
The rehabilitation process (again, following the standard 12 week schedule, spread over 2 years) with the second ABI revealed distinct advantages owing to child A’s age. He was already communicative through sign language and thanks to the first ABI possessed a foundational understanding of the concept of hearing, which significantly facilitated the fitting process. Given the very limited benefit of the first implant and the very high current levels, the rehabilitation of the second implant was done while the left-sided implant was not active anymore. Some contacts of the new implant, while not audibly perceptible to him, induced a tingling sensation in his leg and were subsequently deactivated. The rehabilitation process in the first 8 months has shown promising progress, with all active electrodes providing auditory input. Now, he is very motivated to wear his processor all day, and even motivates his younger brother to do this as well. In Figure 3, the IT-MAIS-NL data of Child A’s second implant are plotted, showing IT-MAIS 11 (on a 40-point scale) and a CAP-score 1 meaning awareness of environmental sounds.
Discussion
Here, we present the successful rehabilitation with ABIs of two brothers with compound heterozygotic pathogenic mutations in the FGF3 gene. The review articleby Jamshidi et al. 3 on the LAMM syndrome showed that in homozygotic cases of both gene mutations (as found in these siblings) labyrinthine aplasia occurred, which was also the case here. The brothers, however, do not exhibit the full LAMM syndrome as both children had just mild and single-sided microtia. However, both have complete labyrinthine aplasia and the bilateral absence of the cochlear nerve, and both turned out to have microdontia (this was not clear at the time of presentation as neonates as they did not have teeth yet). The oligodactyly in child A (for which he underwent a separate surgery) is not considered a part of the syndrome.
The most interesting observation in this study is the fact that the anatomy of the brainstem is abnormal with this gene defect, and therefore is probably a previously unknown aspect the LAMM syndrome. In fact, we could not find any literature on ABI in children with the LAMM syndrome, and to our knowledge, the fact that the CN is located more caudally and deeper into the lateral recess of the fourth ventricle has not been observed before. This finding was substantiated by the good responses to the atypically placed ABI electrode arrays in child B, the second surgery in child A, and, in retrospect, by the fact that the only three electrode contacts with auditory responses in child A’s first implant were located in the caudo-medial portion of the array. We made preoperative MRIs of the whole brain (not shown here), –not only in retrospect-but also could not identify the anatomical differences found during surgery. Although they turned out to be very important determinants of the clinical outcome, the anomalies are, apparently, too subtle to be noticed in imaging.
These cases underscore the importance of individualized care and the potential benefits of reassessing and adjusting treatment strategies based on familial patterns and patient outcomes. For this, it is indispensable to have a multidisciplinary team, which members who collaborate closely in the pre-, peri- and post-operative period. In our centre, we have ample surgical and rehabilitation experience with both CIs and ABIs, as well as with acoustic neuroma/skull base surgery. Thanks to the long-standing collaboration between the members of the surgical team who have jointly performed numerous ABI-surgeries, viz., the ENT- and neurosurgeon, supported by the clinical physicists-audiologists, we were able to identify the pattern of abnormal brainstem anatomy in this syndrome and successfully rehabilitate both children, especially child A after the second surgery.
Conclusion
An abnormally caudal position of the CN in the brainstem was found in two congenitally deaf siblings with cochleovestibular aplasia due to mutations of the FGF3 gene, which is associated with the LAMM syndrome. It is important to be aware of this anatomical abnormality in future cases with this genetic disorder, as correct placement of the electrode is mandatory for a good result with an ABI.
Footnotes
Acknowledgements
The authors wish to thank the parents of the children for their permission to describe and publish their medical history, all team members for their dedication and Dr. R.K. Kalkman for his help in preparing the final draft of the manuscript.