
Abstract
Phosphoglucomutase-1-congenital disorder of glycosylation (PGM1-CDG) (OMIM: 614921) is a rare autosomal recessive inherited metabolic disease caused by the deficiency of the PGM1 enzyme. Like other CDGs, PGM1-CDG has a multisystemic presentation. The most common clinical findings include liver involvement, rhabdomyolysis, hypoglycemia, and cardiac involvement. Phenotypic severity can vary, though cardiac presentation is usually part of the most severe phenotype, often resulting in early death. Unlike the majority of CDGs, PGM1-CDG has a treatment: oral D-galactose (D-gal) supplementation, which significantly improves many aspects of the disorder. Here, we describe five PGM1-CDG patients treated with D-gal and report both on novel clinical symptoms in PGM1-CDG as well as the effects of the D-gal treatment. D-gal resulted in notable clinical improvement in four patients, though the efficacy of treatment varied between the patients. Furthermore, there was a significant improvement or normalization in transferrin glycosylation, liver transaminases and coagulation factors in three patients, creatine kinase (CK) levels in two, while hypoglycemia resolved in two patients. One patient discontinued the treatment due to urinary frequency and lack of clinical improvement. Furthermore, one patient experienced recurrent episodes of rhabdomyolysis and tachycardia even on higher doses of therapy. D-gal also failed to improve the cardiac function, which was initially abnormal in three patients, and remains the biggest challenge in treating PGM1-CDG. Together, our findings expand the phenotype of PGM1-CDG and underline the importance of developing novel therapies that would specifically treat the cardiac phenotype in PGM1-CDG.
Plain Language Summary
PGM1-CDG is a rare genetic disorder that affects glycosylation, an important biochemical process happening in every cell of the body. Because glycosylation is essential for correct functioning of the cells and happens in every tissue and organ, patients with PGM1-CDG can have a variety of symptoms affecting many different organs. Main symptoms include low blood glucose levels, hyperinsulinism, bleeding disorder, liver, muscle, heart problems, and so on. This disorder is usually diagnosed based on the genetic testing, patient’s symptoms, and transferrin glycosylation test, which detects abnormalities in glycosylation in blood. So far, more than 60 patients have been reported. Unlike many genetic disorders, PGM1-CDG has a treatment in the form of a sugar called galactose, which naturally occurs in milk, and can treat many symptoms of the disorder. The patients are advised to take it every day by mouth in the form of powder. Here, we describe five more patients with PGM1-CDG, who were treated with galactose. Each of the patients had novel symptoms and they responded to the treatment differently, which helps us to better understand the disorder and the effects of therapy better. We found that many symptoms improved or normalized; however, some patients experienced persistent symptoms and even adverse events that made them stop treatment. Unfortunately, we did not observe any improvement of heart-related issues. Given that heart issues are the most severe aspect of PGM1-CDG and can result in early death, therapies that target heart issues in PGM1-CDG are still necessary. In conclusion, we describe novel aspects of PGM1-CDG, which will help understand and diagnose the disorder better, and highlight the importance of developing new therapies for this disorder that would specifically treat the heart.
Keywords
Introduction
Congenital disorders of glycosylation (CDGs) are a group of inherited metabolic disorders that affect glycosylation, one of the most important post-translational modifications. 1 The majority of CDGs present as multisystem disease and with varying phenotypic severity. 1 Currently, effective treatments have been described for about 10% of CDGs. 2 One of these CDGs is phosphoglucomutase-1-(PGM1)-CDG (OMIM: 614921), which is treatable with oral D-galactose supplementation (D-gal).3–5 PGM1-CDG is an autosomal recessive inherited metabolic disorder caused by biallelic pathogenic variants in the PGM1 gene, 3 which encodes the PGM1 enzyme. PGM1 interconverts glucose-6-P and glucose-1-P and hence plays an important role in multiple biochemical pathways such as glycolysis, glycogen synthesis, and glycogenolysis.6,7 Due to its involvement in glycogen metabolism, PGM1 deficiency was first characterized as a glycogen storage disorder (GSD XIV).3,8–10 However, abnormal glycosylation with both a endoplasmic reticulum (ER) (CDG type I) and Golgi apparatus (GA) glycosylation defect (CDG type II) was later observed in the blood of the patients, and the disorder was then reclassified as a congenital disorder of glycosylation (PGM1-CDG). 3 This ‘mixed CDG type’, which is characterized by both CDG type I and CDG type II glycosylation pattern, has so far only been described in PGM1-CDG 3 and was not understood at the time the disorder was first described. A recent study, however, has shown that PGM1 deficiency results in a depletion of UDP-glucose and UDP-galactose, which are individually crucial for ER and GA glycosylation, respectively, explaining the mixed CDG phenotype. 11
PGM1-CDG is a multisystemic disorder affecting multiple organ systems and tissues.3,10,12 Most common biochemical findings are elevated transaminases, rhabdomyolysis, and hypoglycemia, which have been reported in more than 65% of the patients. 12 Dysmorphic features such as cleft palate, bifid uvula, and Pierre Robin Sequence (PRS) are present in more than half of the reported patients. 12 Cardiac involvement is also common in PGM1-CDG and usually results in the most severe phenotype and has been reported in about half of PGM1-CDG patients.12–15 The most common cardiac finding is dilated cardiomyopathy (DCM). Electrocardiography (ECG) abnormalities such as sinus tachycardia, prolonged QT interval, and ventricular enlargement were also reported. 12 Restrictive cardiomyopathy was reported in one patient.12,14 Unfortunately, many of the patients with cardiac involvement require cardiac transplantation and premature death due to cardiac complications is not uncommon.12,13,15 PGM1-CDG also frequently presents with coagulation abnormalities, with antithrombin III (ATIII) deficiency as the most frequent finding. 12 Moreover, thrombotic events were reported. 12 Other observed endocrine abnormalities include hypogonadotropic hypogonadism,3,16 decreased levels of growth hormone or insulin-like growth factor 1,3,6,8,9,11,16–21 abnormal thyroid function,10,19,22 abnormal plasma cortisol levels, adrenocorticotropic hormone (ACTH), and gonadotropins (luteinizing hormone LH, follicle stimulating hormone FSH).3,8,11,15,17,19,21–27 Furthermore, the liver is usually affected in PGM1-CDG, with findings including hepatomegaly,11,19,22 steatosis, cholestasis, and fibrosis. 3 Apart from rhabdomyolysis, muscular involvement includes persistently elevated CK, exercise intolerance, fatigability, and muscle weakness. 12 Notably, muscle involvement can also be the only presenting sign of PGM1-CDG, although this has been reported in only a few patients.3,8,23,27 Interestingly, unlike many CDGs, the central nervous system is generally not affected in PGM1-CDG, and cognitive impairment has been reported in just over 20% of the patients. 12 Intellectual disability (ID) and seizures were originally thought to be the consequence of hyperinsulinemic hypoglycemia (HH), though recently it has been implicated that ID and seizures present regardless of glycemic status.22,28 Respiratory system is not commonly affected in PGM1-CDG and the symptoms are usually associated with facial congenital malformations, exercise intolerance, and/or cardiac disease.3,9,15,20,26,27 Gastrointestinal findings are likewise uncommon and usually include feeding difficulties due to facial congenital malformations. 12 Malignant hyperthermia was reported in two patients who have undergone surgery and presented with rhabdomyolysis. 3
The prevalence of PGM1-CDG is unknown and there are currently more than 60 reported patients with a confirmed molecular diagnosis.3,5,10,12–16,19–27,29–32 PGM1-CDG usually presents at birth, 12 and the majority of PGM1-CDG patients are diagnosed in the first years of life. The eldest reported patient up-to-date was 53 years old at the age of diagnosis. 33 Given that effective therapy in the form of D-gal is available for PGM1-CDG, timely diagnosis is crucial. Biochemical methods based on serum transferrin analysis (transferrin isoelectric focusing, or MS based methods) are usually used to detect abnormal glycosylation and help diagnose and monitor CDGs.31,34–37 Based on the transferrin profile, CDGs are allocated into two groups – CDG type I, which affects the ER part of glycosylation, and CDG type II, which affects the GA glycosylation. PGM1-CDG is unique among CDGs as it is so far the only CDG which presents with a mixed CDG type. 3 Although the majority of PGM1-CDG patients present with the mixed CDG type, there are reports of patients with a CDG type I or CDG type II.3,12,31 The appearance of a mixed CDG type is strongly indicative of PGM1-CDG; however, this phenomenon has also been observed in ARCN1 deficient patients during acute illness episodes. 38 Therefore, the final PGM1-CDG diagnosis typically requires molecular genetic testing and can be further confirmed with PGM1 enzymatic activity assay, 12 which usually ranges between 0% and 20 %3,10,11,12 of normal functionality in PGM1-CDG individuals. Unlike some other inborn errors of metabolism, no correlation has been established between the severity of phenotype and the degree of PGM1 enzymatic activity. 10
PGM1-CDG is treatable by D-gal supplementation.3,5,11 The mechanism by which D-gal is able to improve glycosylation is by increasing depleted galactose-1-P and nucleotide sugars (UDP-glucose and UDP-galactose). 11 So far, D-gal has been trialed in more than 20 PGM1-CDG patients, with significant clinical improvement observed in the majority of the treated patients.3,5,11,14,20,21,26,29 Reported D-gal dose ranged between 0.3 and 3 g/kg/day, with the tolerated dose not exceeding 50 g/day.3,5,11,12,14 The biggest improvement in laboratory findings was seen in coagulation parameters and liver transaminases, which normalized in the majority of patients treated with D-gal.3,5,11 D-gal treatment improves transferrin glycosylation, and strategies for transferrin biomarker monitoring indices have been proposed to monitor patient compliance.29,31 Furthermore, D-gal was shown to improve exercise intolerance, hypogonadism, delayed puberty, rhabdomyolysis, and fatigability in some patients.3,5,11,20,21 Hypoglycemia and CK also improved in some patients; 11 however, fluctuating glucose and CK levels have also been observed on D-gal.3,5,11,21,26 Hypoglycemia is usually further managed by frequent feedings and cornstarch, 12 especially in infancy. Nevertheless, D-gal has so far not been successful in treating cardiac presentation in PGM1-CDG. Considering that the cardiac presentation defines the most severe PGM1-CDG phenotype, the development of better therapies that would also improve heart function in PGM1-CDG is still a major unmet clinical need.
Here, we present detailed case reports of five PGM1-CDG patients treated long term with D-gal, each presenting with novel clinical findings and discuss the persisting challenges of treating PGM1-CDG.
Case reports
Five patients with PGM1-CDG were enrolled in the CDG Natural History Study at Mayo Clinic (Mayo Clinic IRB: 19-005187; ClinTrial.gov NCT04199000). In addition, patients were enrolled in the monosaccharide therapy in CDG study (Mayo Clinic IRB: 18-007276, ClinTrial.gov NCT04198987). Written informed consent was obtained from all the patients or their legal guardians. Three of the patients (P1–P3) were previously reported; however, detailed clinical presentation was not provided. 29 Major clinical and biochemical features of all the patients (P1–P5) are described in Table 1 and Table 2.
Biochemical and clinical features of five newly diagnosed PGM1-CDG patients prior to galactose treatment.

CDG, congenital disorder of glycosylation; CK, creatine kinase; DCM, dilated cardiomyopathy; DVT, deep vein thrombosis; GERD: gastroesophageal reflux disease; HTN: hypertension; IGF-1, insulin-like growth factor 1; IGFBP-3, insulin-like growth factor-binding protein 3; LA: left atrial; NR, not reported; PCOS, polycystic ovary syndrome; PGM, phosphoglucomutase; PRS, Pierre Robin sequence; VSD, ventricular septal defect.
First reported in Perales-Clemente et al. 29
At the time of publication.
Detailed biochemical changes recorded before D-gal therapy and at the most recent visit during D-gal treatment.

ALT, alanine transaminase; AST, aspartate transaminase; ATIII, antithrombin III; ATIIII,; CDG, congenital disorder of glycosylation; CDT, carbohydrate deficient transferrin; CK, creatine kinase; N/A, not applicable; NR, not reported.
First reported in Perales-Clemente et al. 29
Patient 1 is a 30-year-old female who has been diagnosed with PGM1-CDG at age of 26. She has been receiving D-gal treatment for more than 3 years. At birth, she was diagnosed with a small posterior midline cleft. Her development was normal apart from a significant speech delay. She had several hypoglycemic episodes in childhood, and she developed exercise intolerance marked by elevated CK and fatigue at the age of 5 years. In addition, she presented with recurrent ear infections and was diagnosed with polycystic ovary syndrome (PCOS) in puberty. She had mild hypothyroidism, a history of migraines and experienced frequent palpitations. Cardiac investigations revealed negative T-waves and a dilated mid-ascending aorta. From 18 years of age, she has been experiencing recurrent episodes of rhabdomyolysis, some of them requiring hospitalization. Liver investigations revealed hepatic steatosis. Laboratory investigations revealed elevated liver enzymes and CK levels while transferrin analysis revealed a mixed CDG type. Genetic testing was positive for two variants in PGM1 (c.313A > T; c.200 T > G, see Table 1), which confirmed PGM1-CDG. Upon diagnosis, D-gal was started at 1 g/kg/day, after which transaminases, coagulation factors and blood glycosylation significantly improved (Figure 1). However, she still experienced episodes of rhabdomyolysis (with CK values as high as > 53,000 U). Furthermore, elevated CK levels and cardiac symptoms persisted on therapy. At age of 28, a loop recorder was implanted after an episode of wide-complex tachycardia. Upon implantation, patient had several episodes of rhabdomyolysis, prolonged muscle pain, nausea, and decreased food intake. Cardiac device has registered two separate episodes of tachycardia since implantation. Recently, a continuous glucose monitor was placed to monitor hypoglycemia for 3 months which revealed morning hypoglycemia. Due to her persisting symptoms, her D-gal dose was increased to 50 g/day.
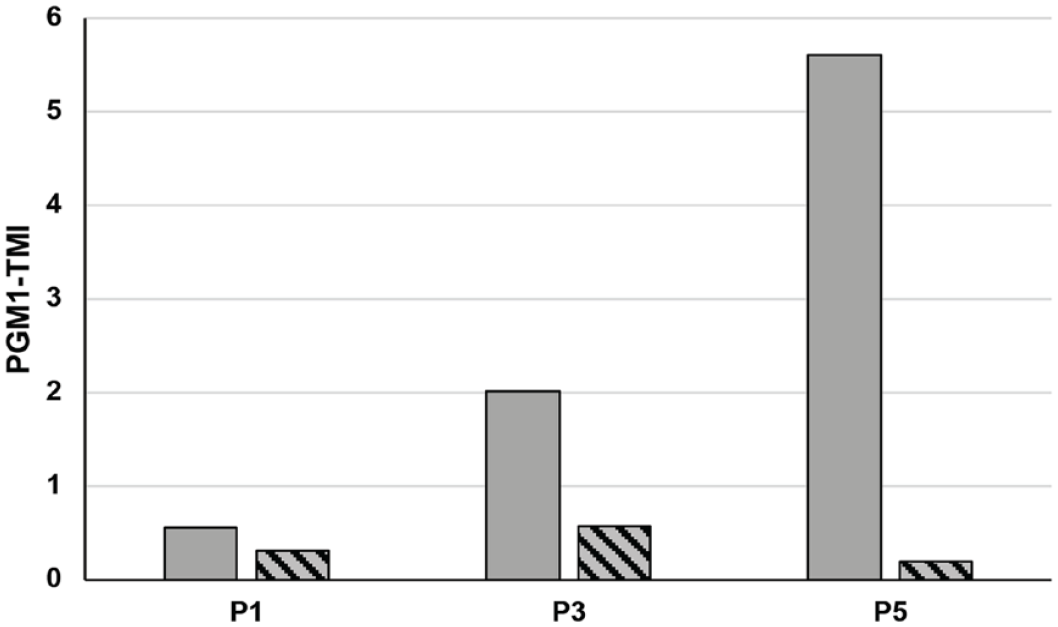
PGM1 monitoring index (PGM1-TMI) 29 of transferrin biomarkers for select patients in the case series.
Patient 2 is a 27-year-old female. She was diagnosed with PGM1-CDG at 24 years of age. At birth she presented with macrocephaly, jaundice, and breathing difficulties due to the PRS and cleft palate. In the first months of life, she had undergone a lip-adhesion surgery, during which she had a cardiac arrest for 7 minutes and required hospitalization for 1.5 months. Later, the patient underwent cleft palate repair without complications. She had notable developmental and growth delay. Vision and hearing were also impaired. Neurologic symptoms included balance issues, memory loss, and encephalopathy. Moreover, the patient exhibited behavior abnormalities and has been taking medication for depression. She also had ID, although no HH was ever reported. Furthermore, fatigability, exercise intolerance, progressive muscle weakness, myalgia, and mild myopathy were present. Mitral valve insufficiency and DCM were also present. Gastrointestinal manifestations included acid reflux, swallowing difficulty and constipation. There were no liver abnormalities. She had acute pancreatitis and deep vein thrombosis. Laboratory investigations showed decreased coagulation factors, and transferrin isoelectric focusing revealed a CDG type I pattern. Whole genome sequencing revealed one known pathogenic and one likely pathogenic variant in PGM1 (c.787G > T; c.988G > C, see Table 1). She has complained of progressive weakness in her hands and worsening balance issues. A single 25 g/day of D-gal has recently been proposed to the patient and D-gal treatment restarted, recently increased to 50 g/day. The patient is currently taking several medications to help manage her symptoms, including mometasone, medroxyprogesterone, betamethasone valerate, digoxin, dulotexine, enalapril, pantoprazole, furosemide, gabapentin, rivaroxaban, sertraline, spironolactone, codeine, Pepcid, and Ativan.
Patient 3 is a 5-year-old male that presented with a two-vessel cord, PRS, cleft palate, and short femurs at birth. He failed the initial hearing test, and he spent the first 40 days of his life in the hospital for mandibular distraction. Mild developmental delay was noted, which was characterized mostly by speech delay, and articulation problems were present. Patient had HH and required frequent overnight feeding through a G-tube. At 2 years of age, the patient experienced an episode of a suspected seizure. However, his electroencephalogram (EEG) was unremarkable. Abnormal involuntary movements and central hypotonia were present. A transthoracic echocardiogram showed mild dilated mitral valve annulus, mild mitral valve insufficiency, severe left atrial dilation, and severe left atrial enlargement. He underwent tonsillectomy and adenoidectomy due to recurrent acute suppurative otitis media. Notable laboratory findings included recurrent abnormal coagulation parameters, elevated liver transaminases, increased CK levels and abnormal glycosylation in blood suggestive of CDG mixed type. Genetic testing revealed two pathogenic variants in PGM1 (c.265G > A; c.988G > C, see Table 1) confirming the diagnosis of PGM1-CDG. At 3 years of age, D-gal has been started (current dose 1.8 g/kg/day in two doses). In addition, cornstarch (2x 1 tbs; approximately 30 g) has been prescribed to further manage hypoglycemia, which has resolved since starting the treatment. CK levels likewise normalized. Moreover, D-gal significantly improved serum transferrin glycosylation and coagulation parameters (Figure 1). Liver transaminases improved but did not normalize. Cardiac presentation did not improve.
Patient 4 is a 3-year-old female, who presented at birth with cleft soft palate, inverted nipples, and breathing and feeding difficulties. She did not pass the newborn hearing screening, and had ear tubes inserted at the time of her cleft palate surgery. Short stature, developmental delay, mild hypotonia, and a (closed) ventricular septal defect (VSD) were present. Skeletal findings included mild anterior wedging involving the L2 vertebral body, mild inferior beaking of the L1 vertebral body, and mild flaring of the ribs posterior laterally with narrowing at the posterior aspect creating an oar-shaped rib appearance. The patient also developed hypoglycemia. In addition, she experienced episodes of non-responsiveness and fixed staring that were suspected to be seizures; however, the EEG was not performed. She had mildly elevated liver transaminases, low coagulation factor activities, and abnormal serum transferrin glycosylation indicative of CDG type II. Genetic testing showed two variants in PGM1 (c.871G > A; c.689duplG, see Table 1). D-gal was started at 1 year of age, and the dose was increased up to 1.5 g/kg/day. During D-gal treatment, patient had an episode of elevated CK. Her blood glucose, coagulation factors, and liver transaminases continued fluctuating on treatment.
Patient 5 is a 2.5-year-old female. The pregnancy was complicated for maternal diabetes, preeclampsia, and hypertension. Emergency C-section was performed at 37 weeks due to poor fetal tone. Cleft palate, micrognathia, and hypoglycemia were present at birth. Right after birth, she was taken to neonatal intensive care unit where she spent 39 days. At 5 weeks of age, a G-tube was placed due to feeding difficulties and poor weight gain. The patient experienced acid reflux, constipation, and recurrent vomiting episodes. At 10 months of age, she was hospitalized for hypoglycemia and started on cornstarch. Abnormal thyroid function, low ferritin levels, chronic rhinitis, elevated transaminases, and coagulation factor abnormalities were also reported. She had a cleft palate repair without complications at 18 months. Cardiac investigations revealed patent foramen ovale. Brain magnetic resonance imaging revealed abnormalities in the splenium of the corpus callosum; EEG and abdominal ultrasound were normal. There was no history of developmental delay except for speech delay which was attributed to cleft palate. Molecular genetic testing revealed two pathogenic variants in PGM1 (c.696_699del; c.125G > A, see Table 1). Serum transferrin glycoform analysis revealed abnormal glycosylation pattern suggestive of PGM1-CDG. She was started on iron supplements and received speech and feeding therapy. She has been taking D-gal for 7 months (current dose 2 g/kg/day), and all of her laboratory findings normalized, except for transaminases which remain marginally elevated. Serum transferrin glycosylation improved on D-gal as well (Figure 1).
Discussion and implications for therapy
Here, we offer a detailed report on the clinical manifestation and outcome of D-gal supplementation in five PGM1-CDG patients, three of which have previously been reported in Perales-Clemente et al. 29 We observed clinical signs that have not been previously reported in PGM1-CDG, expanding the clinical phenotype of the disorder. Novel clinical findings were noted in the cardiac, gastrointestinal, skeletal, and nervous system. Novel cardiac presentations include dilated mid-ascending aorta (P1) and dilated mitral valve annulus (P3). Novel gastrointestinal findings included constipation (P2, P5) and stenosis of the rectum and anus (P3). Other notable findings included encephalopathy (P2), depression (P2), as well as abnormality in splenium in corpus callosum (P5), which were previously not reported. One patient (P1) suffered from migraines, which was previously reported in one patient.3,10 Patient 4 presented with inverted nipples, which are frequent in PMM2-CDG,1,2,39–41 but uncommon in PGM1-CDG. 12 In addition, P4 presented with skeletal abnormalities (including mild anterior wedging involving the L2 vertebral body and mild inferior beaking of the L1 vertebral body, and mild flaring of the ribs posterior laterally with narrowing at the posterior aspect creating an oar shaped rib appearance), which were formerly only reported in one patient. 10 Finally, minimal neurologic involvement was previously considered in PGM1-CDG, with most of the symptoms attributed to be secondary to hypoglycemia. 12 However, recent reports have shown that PGM1-CDG patients can present with neurologic symptoms regardless of hypoglycemia.21,28 Similarly, we observed intellectual disability without hypoglycemia in P2, suggesting that intellectual disability could also be a direct consequence of PGM1 deficiency and/or abnormal glycosylation, as previously suggested. 28
Apart from the novel clinical presentation, we also report here on the effects of D-gal supplementation in our patient cohort. Previously, short term D-gal supplementation has shown positive clinical changes3,5,11,14,21,26 in transaminases, serum transferrin glycosylation, coagulation factors, reproductive hormones, myopathy, while long-term treatment was reported to further stabilize CK levels. 4 Yet, not all aspects of PGM1-CDG are treatable with D-gal and further therapeutic considerations remain. Crucially, so far, there have been no reports on improvement of cardiac function in PGM1-CDG on D-gal. This is essential, as cardiac complications can lead to early death in PGM1-CDG patients and are usually part of the most severe phenotypes. 12
We observed clinical improvement in four of five patients treated with D-gal. No bleeding episodes were observed on therapy. However, D-gal did not resolve all symptoms in our patients, even at higher doses. Blood glycosylation improved or normalized in three of five patients (P1, P3, P5), which we were able to monitor by PGM1 therapy monitoring index (PGM1-TMI) (Figure 1, Supplementary Table 1). In addition, liver transaminases and coagulation factors improved or normalized in three of five patients (P1, P3, P5), CK levels significantly improved in two patients (P3, P5), while hypoglycemia resolved in two patients (P3, P5) (Table 2). It is important to note, though, that two of the pediatric patients received cornstarch or frequent feeding in combination with the D-gal treatment, which might have additionally improved hypoglycemia. P1 experienced recurring, severe episodes of rhabdomyolysis and hypoglycemia while on D-gal. In addition, elevated CK was measured for the first-time during D-gal treatment in P4. P2 experienced adverse effects on therapy, and no clinical improvement was observed, which led to the temporary discontinuation of D-gal. Moreover, three of the patients (P1–P3) had persistent cardiac abnormalities which did not resolve or improve on treatment.
One of the explanations as to why D-gal is ineffective in treating cardiac presentation in PGM1-CDG is that D-gal is unable to treat structural cardiac changes caused by PGM1 deficiency. In the heart, PGM1 protein interacts with ZASP protein, which in case of PGM1 deficiency can result in dilated cardiomyopathy. 42 Yet, this theory does not explain the presence of other cardiac abnormalities seen in PGM1-CDG patients such as long QT interval, ST wave elevation, T wave inversion, and tachycardia. 12 In addition, previous investigations have shown that abnormal glycosylation can negatively affect action potential and channel signaling, resulting in prolonged cardiac action potential and shorter refractory times, providing a link between electrocardial signaling and abnormal glycosylation. 43 In addition, many of the structural proteins, such as collagen, are glycosylated, and its abnormal glycosylation could lead to incorrect collagen fibril assembly. 44 Collagen is one of the main structural heart proteins, which form extracellular matrix (ECM) and provide scaffold for the myofibril formation and play a crucial role in the correct heart function. 45 Therefore, the incorrect glycosylation of these proteins could lead to structural heart abnormalities. In addition, the remodeling capacity of ECM after birth quickly dissipates, limiting the time for the successful therapeutic interventions that could treat these structural abnormalities. 46
Considering D-gal is able to improve glycosylation, 11 these observations suggest that altered glycosylation alone cannot explain the pathobiology of cardiac abnormalities in PGM1-CDG. Other pathomechanisms could, therefore, be at play. For instance, mitochondrial dysfunction and disturbed energy metabolism, which have been frequently associated with cardiac abnormalities,47–52 could contribute to cardiac presentation but have never been studied in the context of PGM1-CDG. Apart from understanding the pathomechanism behind cardiac dysfunction in PGM1-CDG, specific biomarkers related to cardiac abnormalities, which would facilitate development and evaluation of potential therapies, are also lacking.
Therefore, deeper understanding of the cardiac changes elicited by PGM1 deficiency as well as cardiac-specific biomarkers are necessary to propose and trial therapies that would specifically target cardiac presentation in this disorder.
Conclusion
In conclusion, we describe here five PGM1-CDG patients and expand the phenotype of PGM1-CDG and report on the long-term effects of D-gal treatment in these patients. Finally, we pinpoint the limitations of D-gal in treating cardiac symptoms in PGM1-CDG, underlying the importance of developing novel, more efficient, therapies that would treat the cardiac presentation in PGM1-CDG.
Supplemental Material
sj-docx-1-trd-10.1177_26330040221150269 – Supplemental material for Novel insights into the phenotype and long-term D-gal treatment in PGM1-CDG: a case series
Supplemental material, sj-docx-1-trd-10.1177_26330040221150269 for Novel insights into the phenotype and long-term D-gal treatment in PGM1-CDG: a case series by Silvia Radenkovic, Christin Johnsen, Andreas Schulze, Gurnoor Lail, Laura Guilder, Kaitlin Schwartz, Matthew Schultz, Saadet Mercimek-Andrews, Suzanne Boyer and Eva Morava in Therapeutic Advances in Rare Disease
Supplemental Material
sj-xlsx-2-trd-10.1177_26330040221150269 – Supplemental material for Novel insights into the phenotype and long-term D-gal treatment in PGM1-CDG: a case series
Supplemental material, sj-xlsx-2-trd-10.1177_26330040221150269 for Novel insights into the phenotype and long-term D-gal treatment in PGM1-CDG: a case series by Silvia Radenkovic, Christin Johnsen, Andreas Schulze, Gurnoor Lail, Laura Guilder, Kaitlin Schwartz, Matthew Schultz, Saadet Mercimek-Andrews, Suzanne Boyer and Eva Morava in Therapeutic Advances in Rare Disease
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.