
Abstract
Nystagmus is a disorder characterised by uncontrolled, repetitive, to-and-fro movement of the eyes. It can occur as a seemingly isolated disorder but is most commonly the first, or most obvious, feature in a host of ophthalmic and systemic disorders. The number of underlying causes is vast, and recent improvements in the provision of genetic testing have shown that many conditions can include nystagmus as a feature, but that phenotypes overlap significantly. Therefore, an increase in the understanding of the genetic causes of nystagmus has shown that successful novel therapeutics for ‘nystagmus’ can target either specific underlying disorders and mechanisms (aiming to treat the underlying condition as a whole), or a final common pathway (aiming to treat the nystagmus directly).
Plain language summary
Nystagmus is a disorder of eye movement characterised by uncontrolled, to-and-fro movements. It can occur as an isolated disorder, in conditions affecting other parts of the eye, in conditions affecting multiple other parts of the body or secondary to neurological diseases (brain diseases). In recent years, advances in genetic testing methods and increase in genetic testing in healthcare systems have provided a greater understanding of the underlying causes of nystagmus. They have highlighted the bewildering number of genetic causes that can result in what looks like a very similar eye movement disorder.
In recent years, new classes of drugs have been developed for some of the causes of nystagmus, and some new drugs have been developed for other conditions which have the potential to work in certain types of nystagmus. For these reasons, genetics has taught us that identifying new possible treatments for nystagmus can either be dependent on identifying the underlying genetic cause and aiming to treat that, or aiming to treat the nystagmus per se by targeting a final common pathway. A toolkit based on specific treatments for specific conditions is more to have meaningful impact on ‘nystagmus’ than pursuing a panacea based on a ‘one size fits all’ approach.
Introduction to nystagmus genetics
Nystagmus is not a single disorder
Nystagmus is a disorder of eye movement characterised by uncontrolled, repetitive, to-and-fro movements. 1 It is associated with a variety of underlying causes 2 and in itself, causes significant visual loss. 2 Nystagmus in childhood (when considering all forms together) has a total prevalence of at least 1:700–800 children, 3 so it is commonly encountered in eye clinics. It can occur as an acquired disorder (not genetic) or an inherited genetic condition, either in isolation, or associated with a multitude of underlying ocular or systemic disorders. Underlying genetic disorders include those causing poor vision from birth (e.g. major structural abnormalities), retinal disorders (such as inherited retinal dystrophies) or indeed common conditions causing an early loss of binocularity (such as some forms of strabismus). Terminology and classification of nystagmus in the literature and medical texts has been quite complex in the past and the CEMAS classification system 4 has gone some way to reducing confusion in this area. However, different classification systems are still in use, due, in part, to different priorities of those describing it. For example, the CEMAS classification of infantile nystagmus syndrome (INS) describes nystagmus which is often present at 4–6 months of age, beats in the direction of gaze, is commonly associated with null zones, head postures, and dampening on convergence, and shows accelerating slow phases on eye-tracking. This diagnostic category can be very useful for describing what seems to be a final resulting phenotype which responds similarly to surgical treatments and can exclude other forms for nystagmus with specific underlying aetiologies. However, INS can result from many differing underlying causes itself (such as congenital cataract, albinism, etc.) and as such, has limited utility as a singular diagnosis in itself in most clinical settings. For clarity, the following terminology will be used for the rest of this paper:
Advances in genetics are improving diagnosis of nystagmus
For many infants with apparently isolated INS, an underlying cause may become apparent in later life due to some of the associated ocular or systemic features being subtle and due to investigations being scarce or inappropriate for younger infants. Some features of the nystagmus itself (such as direction, onset, intensity, etc.) can help to guide the clinician to an underlying aetiology; however, no hard rules exist, and exceptions are common. For these reasons, in addition to the large number of serious underlying aetiologies, investigation for people with nystagmus has long been directed towards excluding the most urgent conditions (such as acquired neurological disorders, metabolic disease and cancer). Once these have been excluded, diagnostic pathways have tended to vary significantly, meaning that many patients have been left with a diagnosis of ‘idiopathic nystagmus’. With the increasing use of genetic testing in clinical practice and the advent of next-generation sequencing panels, it is now possible to reach an underlying genetic diagnosis for more patients with genetic forms of nystagmus than ever before.5,6 This increase in genetic diagnoses has come largely from gene panels either designed for patients with nystagmus, or from associated panels such as retinal dystrophy panels or ataxia panels that include genes which cause phenotypes that include nystagmus. This increase in genetic diagnoses has identified that a number of conditions known to cause nystagmus in some patients can indeed present as apparently isolated nystagmus and that ‘hypomorphic phenotypes’ are quite common.5,7,8 This has had important implications for investigation pathways and management of patients with genetic forms of nystagmus and has highlighted the necessity of a structured and methodical investigation pathway which may require access to equipment and expertise which is not currently available in all clinical centres. An example of a modern diagnostic workflow for children with nystagmus illustrates that genetic panel testing is now key to an efficient and effective clinical service (Figure 1).
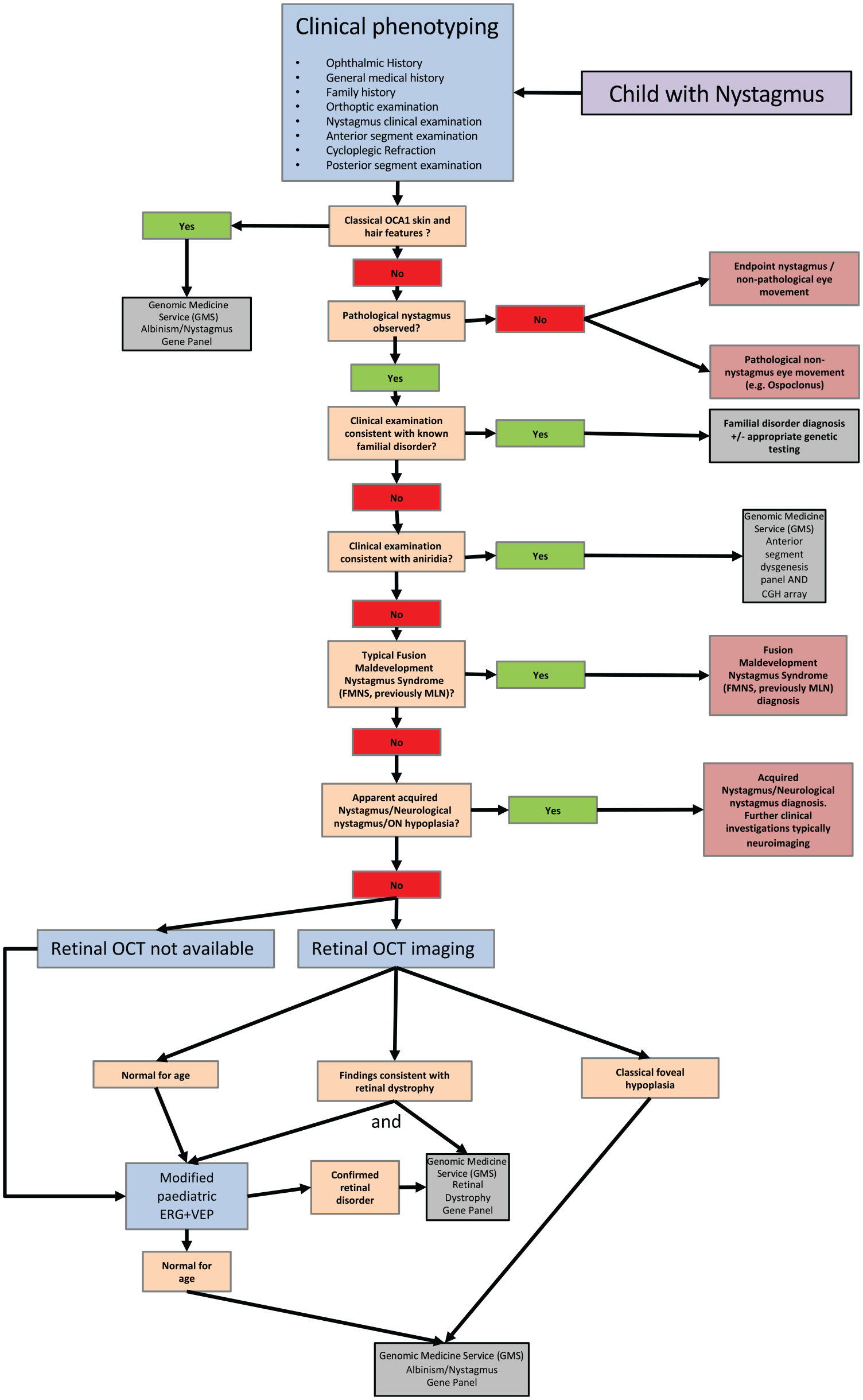
An example diagnostic workflow for children with nystagmus utilising modern UK panel testing. (Adapted from ‘Management of nystagmus in children: a review of the literature and current practice in UK specialist services’. 6 )
Lack of genetic diagnosis for most cases of nystagmus has hampered therapeutic advances
Despite significant advances in diagnostics in some centres,7–9 in the majority of cases worldwide, the underlying cause of nystagmus, even for likely genetic cases, is not identified. This has meant that the development and evaluation of novel therapeutics for conditions which feature nystagmus have often been hampered by small patient numbers or meant that therapeutics have been evaluated on patients without a genetic diagnosis and therefore have been targeted at widely varying underlying aetiologies rather than homogeneous patient groups. It seems that some of the most promising novel therapeutics for people with nystagmus are coming from approaches which have targeted very specific genetic etiologies (e.g. gene therapies for retinal disorders). However, due to the wide variety of underlying conditions which can cause nystagmus, for some cases, the nystagmus is a minor part of the condition (consider a child with severe sight impairment due to Lebers amaurosis) and not the priority target for therapeutic intervention although for others it is the most important debilitating feature (consider those who experience oscillopsia or have IINS).
Advances in the understanding of albinism genetics have shown a complex picture
Through adoption of genetic panel testing in both research and clinical settings it has become apparent that many people with genetic forms of nystagmus have causative genotypes in genes associated with albinism. This is a condition characterised by lack of pigment in the hair, skin and eyes and causes nystagmus in the vast majority. Many of the features can be subtle, and syndromic forms exist and are associated with significant systemic health implications. Improvements in the diagnostic rate for albinism has vastly improved the diagnostic rate for nystagmus as a whole because circa 40% of children with nystagmus and no clear retinal disorder have a form of albinism.5,10 However, studies have also shown that complex genotypes are common,5,7 and many cases with clinically diagnosable albinism still only have a partial genetic diagnosis due to missing heritability and possibly due to further, as yet unidentified, causal variants and additional complex genotypes. A greater understanding of causal genotypes in patients with albinism and nystagmus will help to identify which pathophysiological mechanisms have the most potential for impact when targeting novel therapeutics.
For these reasons it seems likely that advances in therapeutic options for people with nystagmus are likely to be most impactful through the evolution of a toolkit comprising some therapeutics with modest benefit for a large range of underlying aetiologies (independent of pathophysiology) and some therapeutics which are more targeted to underlying genetic aetiologies (targeted at specific pathophysiological mechanisms).
Current approaches to therapeutic intervention for nystagmus based on genetic understanding
Current therapeutic approaches to nystagmus can be broadly divided into generic, non-specific approaches (targeting the nystagmus itself) and targeted approaches for specific underlying causes (targeting the underlying condition). Established generic approaches include treatment of any associated amblyopia, optimising refraction using either spectacle correction and/or contact lenses, dampening of the nystagmus using medications such as gabapentin and memantine, surgery to either dampen the nystagmus, that is, tenotomy, or shift any associated eccentric null point to the primary position, some eye-drop medications 11 as well as providing support and information to affected patients and their families. 6
More recently, based on an increasing understanding of the causes of nystagmus and their underlying molecular and cellular mechanisms, there has been a move towards the development of more targeted treatments for specific conditions, for example, albinism, achromatopsia, Leber congenital amaurosis (LCA) and aniridia. Other conditions are also under study and the following are examples for some specific conditions which cause nystagmus as part of their phenotype and where treatment is targeted towards the underlying disorder which results in nystagmus, rather than the nystagmus per se.
Understanding the molecular and cellular mechanisms responsible for visual impairment in albinism has resulted in the identification of several novel therapeutics
Albinism is a common form of inherited eye disease (1:1000 infants in some populations) that is characterised by ocular and/or cutaneous pigment deficiency, abnormalities of retinal development, nystagmus and visual impairment.3,12–14 Normal pigmentation and retinal development are dependent upon the tyrosinase enzyme (TYR), 15 which catalyses the conversion of tyrosine to dihydroxyphenylalanine (DOPA) and melanin. 16 DOPA regulates retinal development by acting through the ocular albinism type 1 (OA1) receptor to up-regulate pigment epithelial-derived growth factor.17–19 This pathway is disrupted in albinism [e.g. at the level of TYR in oculocutaneous albinism (OCA) or the OA1 receptor in ocular albinism], resulting in abnormal retinal development and visual impairment.20,21 OCA can be divided into two subtypes: OCA-1A, characterised by the complete absence of functional TYR, and OCA-1B, characterised by TYR with limited enzymatic activity.22–25
OCA-1B can be potentially treated with nitisinone, a US Food and Drug Administration approved drug used for the treatment of hereditary tyrosinaemia. It acts by inhibiting 4-hydroxyphenylpyruvate dioxygenase, an enzyme in the tyrosine catabolic pathway, resulting in elevated plasma tyrosine levels.26–29 Stability and function of the defective TYR enzyme in OCA-1B can be improved by increasing the availability of tyrosine. Nitisinone has been shown to increase ocular melanosome melanin content and pigmentation in mice models of OCA-1B, 30 and a pilot clinical trial of nitisinone treatment in patients with OCA-1B resulted in an increase in skin and hair pigmentation and a significant improvement in visual acuity. 31
L-DOPA (levodopa) replacement has shown therapeutic potential in animal models of albinism. Administration of L-DOPA to albino rats prevents misrouting of retinal ganglion cells. 32 Oral replacement of L-DOPA in pregnant albino mouse mothers resulted in accumulation of L-DOPA in the eyes of the foetuses. 18 Expression of tyrosine hydroxylase, in the absence of TYR, in the retinal pigment epithelium of albino mice, rescues the phenotype and visual function, solely due to the presence of L-DOPA alone. 19 In humans, levodopa/carbidopa has been used successfully to improve vision in childhood amblyopia and retinal dystrophies and to treat childhood dystonia.33–35 It remains to be proven if an improvement in visual function can also be achieved in human albinism subtypes using L-DOPA. A recent trial of L-DOPA treatment in OCA failed to demonstrate a significant improvement in best-corrected visual acuity. 36 One of the reasons suggested for the lack of a significant effect in this trial was the age characteristics of the participants, which included adults in whom visual/retinal development was already complete. To date, it has been demonstrated that postnatal retinal development and visual function in albinism can be improved through L-DOPA supplementation, if administered during the critical period of neuroplasticity, in a mouse model of human albinism.37,38 However, L-DOPA is unlikely to be effective in ocular albinism, as it requires and intact OA1 receptor to be effective. 15
Gene replacement therapy has demonstrated potential in Leber congenital amaurosis, achromatopsia and albinism
LCA is a group of inherited retinal degenerations characterised by early-onset severe visual dysfunction and nystagmus. RPE65 gene mutations accounts for up to 10% of autosomal-recessive LCA. RPE65 encodes for the retinoid isomerohydrolase, which is crucial for converting light into electrical signals through the visual cycle. Partial or total loss of function of this protein impairs conversion of all-trans retinal back to 11-cis retinal, preventing the visual cycle from occurring. AAV-mediated gene replacement is proven to be safe and effective for RPE65-LCA, with phase III clinical trial results demonstrating a significant improvement in functional vision (multiluminance mobility testing) in treated patients compared with untreated controls at 1 year. 39 Voretigene neparvovec-rzyl (Luxturna; Spark Therapeutics) is administered via subretinal injection and is the first gene therapy to be approved for the treatment of an inherited retinal disease.
Achromatopsia is an autosomal-recessive disorder (prevalence circa 1:30000–1:80000) associated with infantile nystagmus, loss of cone function and colour discrimination, photophobia and reduced visual acuity. 40 There are at least six known causative mutations. Five of these mutations trigger structural abnormalities in the alpha subunit (CNGA3 41 ) and the beta subunit (CNGB3 42 ) of the cone photoreceptor cyclic nucleotide-gated (CNG) channel, transducin (GNAT243,44) or cone-specific phosphodiesterase (PDE6C45,46) ultimately interfering with the cone phototransduction cascade. The ATF6 gene mutation triggers abnormalities in a transcription factor that activates target genes for the unfolded protein response during endoplasmic reticulum (ER) stress, resulting in increased susceptibility to ER-stress-induced damage and death during cone photoreceptor development. 47 Pre-clinical adeno-associated virus (AAV) vector transgene-mediated (delivered by subretinal injection) rescue of cone function in achromatopsia has been demonstrated in several pre-clinical models, including murine models (Cnga348,49 & Gnat2 50 ), canine models (Cngb3 51 ) and sheep (Cnga352,53).
Proof of principle for AAV-mediated TYR gene transfer in restoring melanogenesis and retinal function has also been demonstrated in a murine model of OCA1. 54
Treatment may be more effective in younger patients with albinism and achromatopsia
It is worth noting that similar to observations in albinism, rescue of retinal function in achromatopsia is age dependent, with older animals gaining little benefit, presumably as a consequence of remodelling of the cone-signalling pathway during development.51,55 This suggests that any potential treatments need to be administered in early childhood to obtain the maximal visual benefit. Interestingly, pre-treatment with intravitreal ciliary neurotrophic factor may improve the responsiveness of older animals to gene therapy, by triggering transient dedifferentiation of the cone photoreceptors. 56 The outcomes of several phase I/II human gene therapy clinical trials for CNGA3 and CNGB3 achromatopsia are currently awaited. 57
Translational readthrough-inducing drugs (TRIDs) show promise as a method of therapeutic nonsense suppression in aniridia
Aniridia is a rare (1:40000–1:100000) pan-ocular disorder characterised by a partial or complete absence of the iris, visual impairment and nystagmus. It is caused by mutations in the PAX6 gene, a gene that plays a critical role in eye development.58,59 Up to 90% of reported PAX6 mutations are attributable to mutations resulting in a premature termination of protein translation and a biologically inactive protein. 60 The nonsense suppression drug 3-[5-(2-fluorophenyl)-1,2,4-oxadiazol-3-yl]benzoic acid (Ataluren, also known as PTC124) bypasses the nonsense mutations restoring functional protein production. 61 Ataluren was initially proven to be effective in models of nonsense-mediated Duchenne muscular dystrophy61,62 and cystic fibrosis.63,64 Proof of concept has been demonstrated for a topical Ataluren (START) formulation in achieving a partial postnatal structural and functional rescue of lens and retinal deficits in a mouse model of human aniridia that incorporates a nonsense mutation. 65 The outcome of a phase II open-label extension study in participants with nonsense mutation aniridia is currently awaited.
Potential future approaches to therapeutic intervention for nystagmus based on genetic understanding
Translational readthrough-inducing drugs (TRIDs) for nystagmus
As detailed above, some exciting preliminary work has been done to study the effect of translational readthrough-inducing drugs (TRIDs) for aniridia which is a common cause of nystagmus. However, nystagmus is seen in such a large range of underlying genetic conditions, that making a large impact on nystagmus as a singular group, using TRIDs, depends on the frequency of nonsense mutations in each underlying condition. Approximately 12% 66 of inherited genetic disorders are caused by in-frame nonsense mutations (which might be amenable to TRID rescue). However, this percentage varies significantly between different underlying conditions that lead to nystagmus. Furthermore, it has become clear that the efficacy of TRIDs depends on the specific nonsense variant and local environment meaning that different nonsense mutations, in different genes which cause nystagmus are likely to respond in different ways to TRID rescue. 67 With the discovery of new and derivative molecules with TRID efficacy, it seems possible that a variety of therapeutics may be developed from this drug class with relevance to nystagmus; however, this is likely to comprise a broad toolkit rather than a single therapy for a variety of different genetic causes of nystagmus.
Targeting therapeutics based on common disease-causing variants in specific genes
An alternative approach to seeking therapeutics which correct specific forms of genetic variant is to seek the most common specific variants for an individual condition, study the specific pathophysiology associated with that/those variant(s) and seek very specific, targeted therapeutic intervention strategies. This has been common for conditions for which specific mutations are common (such as the p.Phe508del CFTR gene variant in cystic fibrosis). However, this is clearly less appropriate for conditions in which common causative mutations have not been identified and also depends on the percentage of cases caused by the ‘common’ mutations. This has potential relevance to nystagmus through the identification of common variants which contribute to causal genotypes in albinism such as the R402Q variant in the TYR gene which is part of a complex genotype which seems likely to cause OCA1 OCA type 1 (OCA1). 5 This variant is a very common variant in western populations (circa one in five people) and seems to contribute to causal genotypes in this condition through acting in consort with another, even more common variant in this gene (S192Y), when occurring in trans with a more deleterious TYR variant. 7 Due to the frequency with which this variant is thought to contribute to causal genotypes in OCA1, targeting this specific mutation could have a significant impact on ‘nystagmus’ as a whole.
Targeting therapeutics on pathophysiological mechanisms common to multiple variants in the same gene
This approach seeks to exploit common mechanisms of disease which arise from many different genetic variants in the same gene. For example, for both OCA1 and OCA3, a common underlying pathophysiological mechanism seems to be retention of the OCA1 or OCA3 proteins in the ER (ER retention)68–71 and subsequent loss of functional protein due to ER-associated degradation (ERAD). This process is also seen in other disorders such as some forms of cystic fibrosis and a class of ERAD-inhibitor compounds has been used to try to rescue this common pathological mechanism. Therefore, it is possible that this approach may show some promise for albinism, which is one of the most common causes of nystagmus.
Summary
In summary, the recent advances in understanding the genetic causes of nystagmus has shown that a bewildering variety of underlying genetic disorders can lead to very similar-looking nystagmus phenotypes.5–7,10,72 Indeed, the range of underlying genetic causes is often only narrowed by specific phenotyping methods which are not widely available. 6 For this reason, new diagnostic pathways and workflows are being developed and are helping to provide diagnosis to more and more patients.6,10 This increase in molecular diagnoses has led to the discovery of the most common causative genes, the most common mutations in those genes, the identification of complex genotypes which are still only partially understood, and a greater understanding of pathophysiological mechanisms common to multiple causative mutations in single genes. These insights have identified a variety of potential specific targets for future therapeutic research aiming to treat the underlying condition, in addition to other therapeutic approaches aiming to treat the nystagmus per se. They have also collectively demonstrated that identifying a single treatment for all nystagmus-causing conditions is less likely to bear fruit than the progressive evolution of a therapeutic toolkit based on specific treatments (aimed at treating underlying conditions) for specific molecular diagnosis, combined with more generic treatments for the final common pathway, in some cases.
Footnotes
Author contributions
Jay Self: conceptualisation; formal analysis; methodology; writing: original draft; writing: review and editing.
Helena Lee: conceptualisation; formal analysis; writing: original draft; writing: review and editing.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethics statement
Ethical approval and informed consent was not required for this review.