
Abstract
Worldwide 20,000–40,000 children with congenital or childhood cataract are born every year with varying degrees and patterns of lens opacification with a broad aetiology. In most cases of bilateral cataract, a causative genetic mutation can be identified, with autosomal dominant inheritance being most common in 44% of cases. Variants in genes involve lens-specific proteins or those that regulate eye development, thus giving rise to other associated ocular abnormalities. Approximately 15% of cases have systemic features, hence paediatric input is essential to minimise comorbidities and support overall development of children at high risk of visual impairment. In some metabolic conditions, congenital cataract may be the presenting sign, and therefore prompt diagnosis is important where there is an available treatment. Multidisciplinary management of children is essential, including ophthalmic surgeons, orthoptists, paediatricians, geneticists and genetic counsellors, and should extend beyond the medical team to include school and local paediatric visual support services. Early surgery and close follow up in ophthalmology is important to optimise visual potential and prevent amblyopia. Routine genetic testing is essential for the complete clinical management of patients, with next-generation sequencing of 115 genes shown to expedite molecular diagnosis, streamline care pathways and inform genetic counselling and reproductive options for the future.
Lay abstract
Cataract is a clouding of the lens in the eye. Cataract occurring in children has many different causes, which may include infections passed from mother to child during pregnancy, trauma, medications and exposure to radiation. In most cases of cataract occurring in both eyes, a genetic cause can be found which may be inherited from parents or occur sporadically in the developing baby itself while in the womb. Cataracts may occur on their own, with other eye conditions or be present with other disorders in the body as part of a syndrome. Genetic testing is important for all children with cataract as it can provide valuable information about cause, inheritance and risk to further children and signpost any other features of the disease in the rest of the body, permitting the assembly of the correct multidisciplinary care team. Genetic testing currently involves screening for mutations in 115 genes already known to cause cataract and has been shown to expedite diagnosis and help better manage children. Genetic counselling services can support families in understanding their diagnosis and inform future family planning. In order to optimise vision, early surgery for cataract in children is important. This is because the brain is still developing and an unobstructed pathway for light to reach the back of the eye is required for normal visual development. Any obstruction (such as cataract) if left untreated may lead to permanent sight impairment or blindness, even if it is removed later. A multidisciplinary team involved in the care of a child with cataract should include ophthalmic surgeons, orthoptists, paediatricians, geneticists and genetic counsellors, and should extend beyond the medical team to include school and local child visual support services. They will help to diagnose and manage systemic conditions, optimise vision potential and help patients and their families access best supportive care.
Introduction
Worldwide, 20,000–40,000 children with congenital or childhood cataract are born every year, and there are an estimated 200,000 children blind from bilateral cataract. 1 In the UK, childhood cataract affects 2.5–3.5 per 10,000 children, with most occurring within the first year of life. 2 Genetic mutations account for the majority of cases of bilateral cataract, and the most frequent mode of inheritance is autosomal dominant seen in 44% of families. 3 Important environmental factors to consider include congenital infections such as toxoplasma, syphilis, varicella zoster, parvovirus B19, coxsackievirus, rubella, cytomegalovirus (CMV) and herpes simplex virus I and II (TORCH). Trauma and iatrogenic causes such as medications and radiation exposure are also relevant but rare in this age group. Inherited congenital cataract may occur in isolation (70%), with other ocular abnormalities (complex) (15%) or form part of a syndrome (15%). 4 Because a wide range of conditions is associated with congenital cataract, often many investigations are performed to identify an underlying cause. There is variability in the investigative pathways of paediatric cataract patients, with some not undergoing any testing despite a family history, or inappropriate and inefficient use of screening tests. 5 Implementation of genetic testing using next-generation sequencing has provided evidence of streamlining this process. 6
Surgery for paediatric cataract has many considerations and early interdisciplinary management is important for good long-term visual outcomes. General paediatric involvement is essential to aid diagnosis and management of children presenting with systemic features and in the management of vision in the developing child as well as supporting families in challenges that may present throughout childhood. There is often significant responsibility placed on parents who have a critical role in adherence to intensive treatment – for example, with patching or frequent administration of eye drops post-operatively and multiple hospital visits.
Aetiology and classification
The human lens is formed in utero from surface ectodermal cells, which thicken to form the lens placode and then invaginate and pinch off to form the lens vesicle by day 33 of gestation.7,8 The posterior epithelial cells elongate anteriorly to occlude the lumen of the vesicle and form the primary lens fibres. Only the anterior epithelial cells at the equatorial regions of the lens divide to form secondary lens fibres, and this continues throughout life (Figure 1). 9
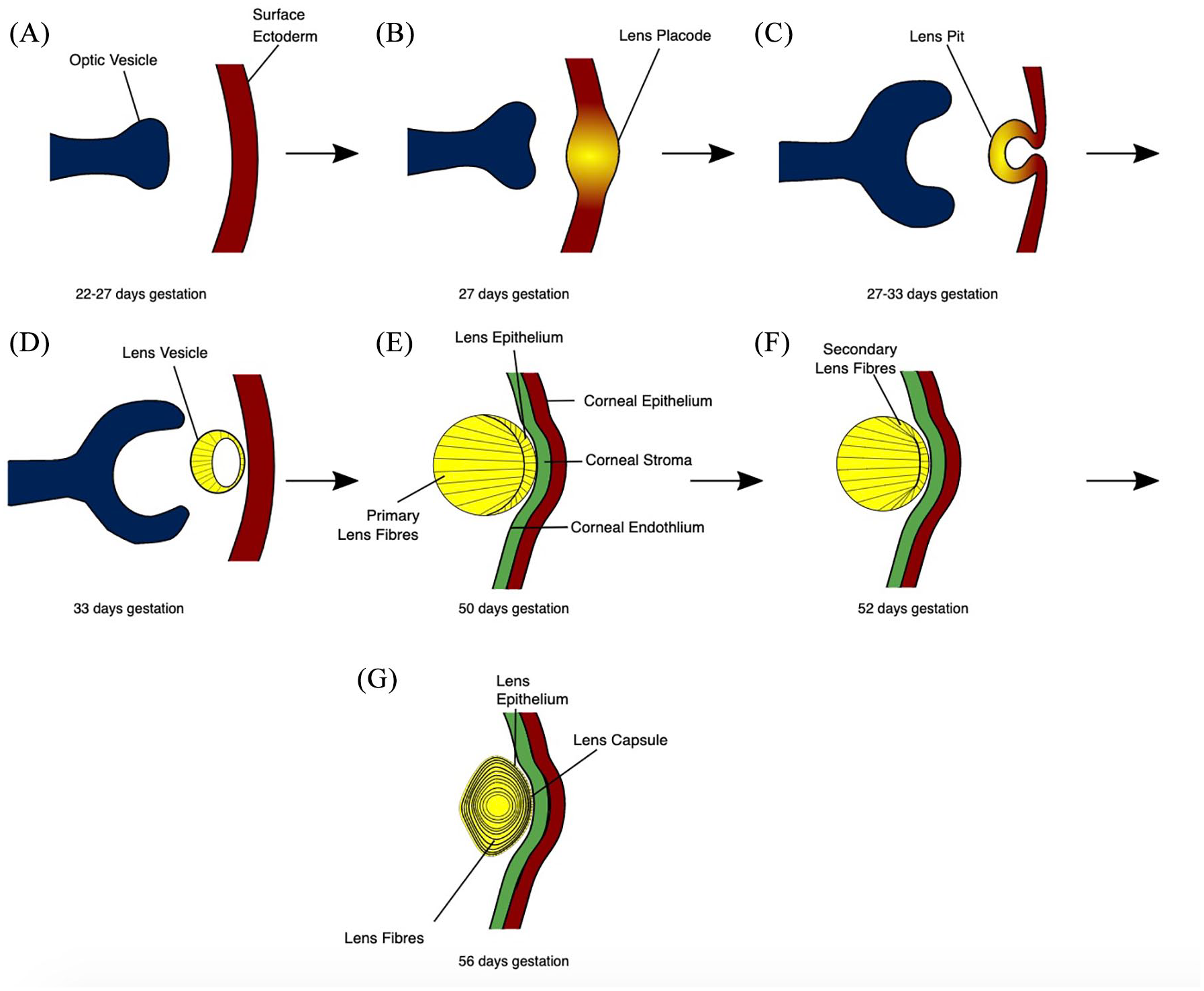
The stages of human lens development. (a) The preplacodal region develops within the surface ectoderm overlying the optic pit/groove. (b) Signalling from the optic vesicle stimulates the preplacodal region to thicken to form the lens placode. (c) Cell proliferation at the lens placode, in addition to morphological changes including apical constriction, result in the formation of a lens pit. (d) Detachment of the lens vesicle from the surface ectoderm, which is rebuilt to later form the corneal epithelium. (e) Posterior epithelial cells elongate anteriorly to occlude the lumen of the lens vesicle. Maturation of the cornea into the epithelium, stroma and endothelium. (f) Epithelial cells at the equatorial regions form secondary lens fibre cells, which elongate anteriorly and posteriorly, encircling the primary lens fibres.
Cataract develops due to disruption of the normal lens protein structure or function, resulting in opacity. This may occur as a result of stressors applied to lens proteins including those acquired in utero or during childhood. However, a significant proportion is due to mutations in the genes encoding lens proteins that directly affect their role within the lens. 8 Various congenital cataract morphology have been described, including nuclear, posterior/anterior polar, pulverulent lamellar, posterior subcapsular, cerulean or blue dot cataract (Figure 2).

Slit-lamp presentation of inherited cataract phenotypes: (a) anterior polar cataract; (b) blue dot (cerulean) cataract; (c) posterior subcapsular cataract; (d) nuclear cataract (retroillumination, kindly donated by Professor Ian Christopher Lloyd).
Cataract can be categorised according to age of presentation. True congenital cataract presents at birth, while cataract that develops later in childhood is described in various ways (e.g. developmental, infantile, juvenile) with different criteria for inclusion in these classifications.10–12 Categorisation in this way can be challenging, as cataract may exist at birth but not be identified until later, which is important in countries where delayed presentation is an issue. 13
Inherited cataract
The proportion of cataract due to genetic mutations is likely higher than we think because many patients are not tested and there are more cataract causing genes to be discovered. It is possible to identify a genetic mutation in up to 90% of cases of bilateral cataract with current genetic testing. 3 There is substantial genetic and phenotypic heterogeneity with significant intra- and inter-familial variability. Mutations in genes encoding lens proteins can also demonstrate pleiotropic effects – for example, NHS gene, which causes Nance–Horan syndrome has a complex pattern of temporally and spatially regulated gene expression involving the development of ocular, craniofacial and neural tissue.14,15 For some cataract-associated genes, the resulting phenotype can vary widely depending on the localisation of the respective nucleotide change. The LSS gene encodes lanosterol synthase, which is an enzyme involved in the biosynthesis of cholesterol in the body. Romano et al. suggested that autosomal recessive mutations occurring towards the N-terminus of this gene are associated with hair loss and those at the C-terminus are associated with lens defects. 16 This theory is consolidated by Chen et al., who reported a mixed phenotype in an individual with baldness, absent eyebrows and congenital cataracts with two heterozygous mutations occurring in LSS c.1025T>G, p.(Ile342Ser) and c.1887G>T, p.(Trp629Cys) near the N- and C-terminal regions respectively.16,17
Non-syndromic congenital cataract
The main lens proteins associated with congenital cataract include crystallins, membrane proteins, cytoskeletal structural proteins and transcription factors. Some of these genes are also involved in the development of other ocular structures; hence in approximately 15% of cases mutations can lead to associated eye abnormalities. 11
Crystallins
α, β and γ-crystallins are the main structural refractive protein of the human lens; thus mutations in this family of genes result in opacity. In addition, α-crystallins behave like molecular chaperones, where they assist in the folding or unfolding of other damaged or denatured proteins such as β and γ-crystallins, and can inhibit apoptosis. 18 These functions are critical for maintaining lens transparency. Approximately 50% of non-syndromic hereditary cataract are due to mutations in genes coding for crystallin proteins with over 100 mutations, mostly missense, encoding 12 different human α, β and γ-crystallin genes (Table 1). 19
Genes associated with isolated cataract. Detailed phenotype data sourced from Genomics England PanelApp and OMIM cataract databases and associated relevant publications.

AD, Autosomal dominant; AR, autosomal recessive; XD, x-linked dominant.
Cytoskeletal structural proteins
Beaded filament structural proteins (BFSPs) are a type of intermediate filament protein that are expressed in various cells of the body, such as epithelial, glial and muscle cells. BFSP1 and BFSP2 are genes encoding Filensin and Phakinin, respectively, which are proteins expressed exclusively in the lens. They are intimately associated with crystallins, where they form part of a highly organised cytoskeleton and are also thought to have an important role in lens development and differentiation. 20 Autosomal recessive BFSP2 mutations cause cataract, but interestingly it has been suggested that heterozygous carriers of these mutations may be predisposed to develop age-related cataract and myopia (Table 2). 20
Genetic mutations associated with cataract and other ocular malformations. Detailed phenotype data sourced from Genomics England PanelApp and OMIM cataract databases and associated relevant publications.

AD, Autosomal dominant; AR, autosomal recessive; XD, x-linked dominant; ASD, anterior segment dysgenesis.
Membrane proteins
The lens is an avascular structure and therefore membrane proteins have an important role in maintenance and metabolic homeostasis. Genetic mutations in over 10 membrane protein genes lead to inappropriate transport of ions, solutes and water between cells in the human lens (Table 1). Gap junction channels are made up of two connexons composed of six subunits called connexins. 21 Connexins have an important role in lens microcirculation, particularly in the supply of metabolites and nutrients towards the centre of the lens, and outward flow of unwanted ions and by-products to the periphery. 22 They are able to form functional hemichannels in a variety of species. Three particular cataract-causing mutations result in increased hemichannel activity: GJA3 (encoding human lens connexin 46) c.427G>A, p.(Gly143Arg) and c.56C>T, p.(Thr19Met), and GJA8 (encoding connexin 50) c.137G>T, p. (Gly46Val). Two mutations in GJA8 c.827C,T, p.(Ser276Phe) and c.131T>C, p.(Val44Ala) decrease hemichannel activity and have also been implicated in the formation of cataract. 23 Mutations in these genes are most commonly associated with zonular and nuclear pulverulent cataracts with varying progression.23–25 These mutations are most frequently missense, and GJA8 is also associated with microcornea.26,27
Mutations in genes encoding major intrinsic proteins account for 5% of all inherited cataract. 28 Aquaporin-0 is the most expressed membrane protein in the human lens and variants are inherited in an autosomal dominant pattern producing a variety of cataract phenotypes (Table 1).29,30 It functions primarily as a water channel, but recent research in mouse models suggests that it may also modulate gap junctions in the presence of BFSPs. 31 Lens intrinsic membrane protein-2 (LIM2) is the gene responsible for a membrane protein with four transmembrane domains called MP19, which contribute to lens transparency.32–34 Three LIM2 missense mutations have been described that cause autosomal recessive cataract (1) c.313T>G, p.(Phe105Val) mutation associated with cortical cataract; (2) c.587G>A, p.(Gly154Glu) mutation in a family causing autosomal recessive cataract; and (3) c.233G>A, p.(Gly78Asp) mutation in a consanguineous Pakistani family with nuclear cataracts.35,36
Other proteins working at the membrane include EPHA2 and DNMBP. EPHA2 encodes a membrane-bound protein tyrosine kinase, 37 and mutations have been shown to account for approximately 5% of inherited cataracts in the Australian population. 38 Most known causative mutations occur in the sterile-α-motif region of the molecule that affects the structure of the EPHA2 protein and impairs cell migration in human and mouse fibroblast lens epithelial cells. 39 Mislocalisation of two mutant proteins away from the cell membrane has also been implicated in cataractogenesis. 37 EPHA2 mutations are most often associated with autosomal dominant posterior polar cataract. DNMBP encodes a protein that regulates the configurations of cell junctions through binding to tight junction protein 1. Biallelic loss-of-function variants have been shown to result in autosomal recessive cataract with other ocular features, including pupil abnormalities, strabismus and nystagmus. 40
Transcription factors
Heat shock transcription factor 4 (HSF4) protects lens proteins from cell stressors and has a regulatory role in the differentiation of lens fibre cells. 41 Mutations in HSF4 most often produce lamellar cataract, which can be present at birth or develop in early childhood, and can be inherited in a dominant or recessive mode. Of the 16 mutations described so far, 13 are missense and 11 occur in the protein’s DNA binding domain; these are autosomal dominant, suggesting this region is essential for normal protein function. 42
Similarly, MAF encodes a transcription factor containing the basic-leucine zipper (bZIP) domain in which 7 of the 18 known mutations are associated with ocular defects such as iris coloboma, glaucoma, microcornea, microphthalmia, myopia, nystagmus and Peters anomaly. 42 This suggests the bZIP domain has a critical role in eye development. Mutations in the N-terminal upstream of a transactivation domain encoding region of the MAF gene are associated with Ayme-Gripp syndrome (cataract, reduced growth, sensorineural hearing loss, learning disability, brachycephaly and seizures) and Asperger syndrome (Table 3). 42
Genetic mutations associated with syndromic cataract. Detailed phenotype data from Genomics England PanelApp and OMIM cataract databases and associated relevant publications.

AD, Autosomal dominant; AR, autosomal recessive; XD, x-linked dominant; XR, x-linked recessive; IC, isolated cases.
FOXE3 is one of a family of genes encoding transcription factors with a ‘forkhead’ DNA binding domain and has a role in lens embryology. Twenty-four unique mutations have been shown to affect various ocular structures, including cornea, iris, lens and retina (Table 2). 42 Most mutations are missense and 10 occur in the forkhead DNA binding domain, resulting in defective or non-functional protein. Pituitary homeobox 3 (PITX3) proteins are transcription factors that have been shown to play an important role in controlling lens epithelial cell maintenance and fibre cell differentiation in mice. 43 They contain the homeobox and the otp, aristaless, rax (OAR) domains; however, surprisingly no cataract-causing mutations have been described in either domain. Mutations in PITX3 genes most often cause posterior polar cataract with or without anterior segment dysgenesis (ASD), but can result in other ocular defects such as corneal opacity, microphthalmia, microcornea, nystagmus and glaucoma. 42
Syndromic congenital cataract
Syndromic congenital cataract manifests as a result of various types of mutations, including chromosomal abnormalities, loss-of-heterozygosity, mitochondrial disorders, triplet repeat disorders and more complex genetic disorders such as diabetes. 35 Cataracts show phenotypic variability and are associated with a myriad of systemic dysmorphic features. 44
Chromosomal abnormalities featuring cataract include those with an extra copy of chromosome 21 and 13 causing Down’s syndrome (trisomy 21) and Patau syndrome (trisomy 13) respectively. Children with trisomy 21 are born with characteristic features and are prone to various health problems (Table 3), including ocular abnormalities such as cataract, upslanting palpebral fissures, astigmatism, iris abnormalities, strabismus, lacrimal system obstruction, blepharitis, retinal abnormalities, hypermetropia, amblyopia, nystagmus and myopia. 45 Cataract has been found in 15% of cases of children with trisomy 21. 46 Children with trisomy 13 commonly present with cataract as well as microphthalmia and ocular coloboma; however, they rarely live more than a few days or weeks due to multiple severe congenital abnormalities involving the craniofacial, musculoskeletal, cardiac, abdominal and nervous system. 47
Multiple X-linked disorders demonstrate congenital cataracts (Table 3). Lowe syndrome is X-linked recessive, characterised by a triad of features including dense congenital cataracts, intellectual disability and proximal tubular dysfunction. It is caused by mutations in the OCRL gene that encodes inositol polyphosphate 5-phosphatase OCRL-1 and is involved in a large spectrum of intracellular processes, some of which affect actin stress fibres, polymerisation and distribution, which are likely to be responsible for the clinical phenotype. 48 In the neonatal period, hypotonia and cataracts may be seen but the condition may not be diagnosed until later, when renal dysfunction becomes apparent. 49
Norrie disease is an X-linked recessive disorder caused by mutations in the NDP gene, which encodes Norrin. There are genotype–phenotype correlations with missense mutations in exon 3 resulting in less severe phenotypes than nonsense mutations. 50 Male children often present in the first few weeks of life with blindness due to bilateral retrolental vascularised masses with normal-sized eyes. Shallow anterior chamber, iris atrophy, cataracts and corneal opacity are common features, with eyes becoming phthisical over time. 51
Male children with inherited Nance–Horan syndrome have typical facial dysmorphic features (long face, large ears, prominent nose) as well as dental and ocular abnormalities including cataract and developmental delay (Table 3). 52 Interestingly, female carrier children may demonstrate a milder phenotype with Y-sutural lens opacities with cortical ‘riders’ and milder facial and dental abnormalities. 53 Loss-of-function mutations in the NHS gene result in the syndromic disease; however, translocations such as duplications can cause non-syndromic isolated cataract. 52 It has been suggested that this protein could be involved in the regulation of tight junction proteins or in the regulation of actin remodelling and maintaining cell morphology, but the function of this protein remains unknown.54,55
Myotonic dystrophy 1 is also inherited autosomal dominantly but occurs as a result of a cytosine–thymine–guanine (CTG) repeat disorder and is associated with a variety of ocular signs such as cataract, retinal degeneration, low intraocular pressure, eyelid ptosis, epiphora, corneal lesions, extraocular myotonia and weakness and abnormal central control of eye movement. 56 Cataracts are often non-specific and appear as punctate opacities. 57 Neurofibromatosis type 2 is caused by autosomal dominantly expressed mutations in the NF2 gene. It classically presents in adulthood with bilateral vestibular schwannoma (VS); however, in children it most often presents with small posterior capsular or cortical edge cataracts as well as non-VS neurological signs and subtle skin tumours. 58
Metabolic disorders are an important differential in the cause of congenital cataract, and these are often inherited in an autosomal recessive pattern. Systemic presentations may be mild with cataract being the initial sign. Zellweger spectrum disorder (ZSD) is an autosomal recessive metabolic peroxisome biogenesis disorder presenting with severe features in childhood that result in early death (Table 3); milder phenotypes include neonatal adrenoleukodystrophy and infantile Refsum disease. 59 It is caused by mutations in PEX genes that encode proteins required for peroxisome biogenesis. 60
Several rare autosomal recessive conditions are associated with the development of cataract. Rothmund–Thompson syndrome is a rare disorder with only 300 known cases. Two-thirds of cases are caused by a mutation in the RECOL4 gene that is involved in DNA replication and repair, thus leading to widespread changes in skin, connective tissue and bone. There is an increased incidence of cancer in childhood and adolescence, in particular bone malignancies (osteosarcomas) and skin cancers, including squamous and basal cell carcinomas. Cockayne syndrome is a rare autosomal recessive disorder presenting with varying severity. Genetic mutations in ERCC6 or ERCC8 affect the repair of mitochondria and DNA damage – for example, due to environmental stressors such as sunlight leading to photosensitivity. 61 Mutations in the CYP27A1 gene lead to an accumulation of cholesterol and cholestenol in tissues and cause cerebrotendinous xanthomatosis (CTX). 62 Developmental cataract is often an early ocular sign of this condition, along with systemic features such as infantile diarrhoea and tendon xanthomas; however, often a diagnosis of CTX is not made until adulthood when patients present with a variety of progressive neurological features. Early recognition and treatment with daily chenodeoxycholic acid can halt or even reverse severe neurological complications and improve prognosis. 63 Galactosaemia, a rare inherited disorder of galactose metabolism screened for at birth in the UK, is caused by mutations in genes that encode enzymes of galactose. Galactokinase deficiency and classic galactosaemia are caused by mutations in the genes of galactokinase (GALK1) and galactose-1-phosphate (GALT), respectively (Table 3). Nearly 200 mutations in the GALT gene have been found and presentation is often severe in the first few weeks of life as infants are exposed to breast/formula milk. 64 Accumulation of galactitol in the lens causes a characteristic ‘oil droplet’ cataract. Anterior and posterior subcapsular cataract may also occur. 65 Early recognition and initiation of a galactose-free diet can reverse lens clouding and prevent fatal systemic consequences. 66 Galactokinase deficiency is seen as a mild form of galactosaemia and paediatric cataract is often the major clinical feature. 67
Acquired causes
Congenital infections
Rubella is the most common congenital infection causing cataract worldwide. 68 Maternal rubella infection during pregnancy puts the foetus at risk of developing a clinical rubella syndrome comprising a triad of deafness, cataract and cardiac disease. Rubella infection continues to be a public health problem in countries that do not have adequate national immunisation programmes.69,70 Other infections causing cataract include toxoplasmosis, CMV and herpes simplex (HSV I and II). It is common practice to screen for these infections when investigating the cause of paediatric cataract using the TORCH screen, but many factors can affect the positivity of the test, including maternal antibodies in neonates. 71 There is some debate over the use of screening in routine practice and a move towards using specific tests for individual cases based on maternal antenatal history, child comorbidities and vaccination status. 72
Trauma
Traumatic cataract makes up a significant proportion of childhood cataract. 73 It has different considerations in terms of its presentation and management compared to true congenital cataract due to its unilaterality and potential damage to other ocular structures, such as angle recession leading to secondary glaucoma. Surgical management can be more complex but with overall less dense amblyopia and better chance of visual rehabilitation. 74 Common objects causing trauma vary globally, with bow and arrows being the most common cause of ocular trauma in India, followed by sticks, stones and thorns, which are more common in East Africa.74,75
Other common causes of cataract in children
Uveitic cataract and steroid-related cataract
Anterior uveitis is defined as inflammation of the iris and ciliary body. Juvenile idiopathic arthritis (JIA) associated uveitis is the leading cause of ocular morbidity in paediatric patients with uveitis and is associated with HLAB27 positivity. JIA describes a group of arthridities presenting in childhood, and uveitis is an extra-articular feature present in 11–13% of patients. It is more prevalent in females, those with oligoarthritis and positive antinuclear antibodies. Cataract occurring in the context of anterior uveitis may occur as a result of disease-associated inflammation or the prolonged use of steroids in its management, giving rise to deprivation amblyopia and visual loss.76,77 Most commonly steroids produce a posterior subcapsular cataract, and although the precise mechanism is unknown, glucocorticoids are thought to have an important role in the transcription of genes in lens epithelial cells. 78
Radiation cataract
Exposure to radiation can cause the development of cataract. Children undergoing radiotherapy for treatment of childhood cancers such as leukaemia, central nervous system tumours, lymphomas, kidney cancer, neuroblastoma, soft tissue and bone sarcomas are therefore at risk of developing cataract. At higher doses of radiation – that is, above 3 Gy – cataract develops more quickly; however, more recent evidence suggests it can occur even at lower doses of radiation of 0.5 Gy, with the likelihood following a linear dose response. 1 Doses of radiation cannot easily be reduced in the treatment of cancer, but measures can be taken to protect the eye and lens in high-risk patients, such as accurate radiation dose estimates and shielding. 1
Management
Paediatric cataract surgery
The management of cataract presenting in children is more complicated than that of adult patients. Primarily the risks of performing early surgery need to be balanced with facilitating the best possible visual development in children during an amblyogenic period. In children, cataract surgery with or without anterior capsulotomy, posterior capsulotomy and anterior vitrectomy are performed. 79 Intraocular lens (IOL) insertion is another point of contention. Findings from 5-year follow-up studies suggest that for children younger than 2 years old, inserting an IOL often results in similar post-operative vision when compared with contact lens only, it does not protect against post-operative glaucoma and results in additional requirement for intraocular surgery.80,81 Therefore, the recommendation is that children are left aphakic, and certainly those less than 6 months of age, until further evidence is presented. 82 Selecting an IOL is difficult as the eye is growing and power calculations are more challenging because of difficulty attaining accurate measurements in the child. It is recommended that cataract should be extracted at 6 weeks of age for unilateral cataract and between 6 and 8 weeks of age in bilateral cataract. 80 Rigorous post-operative care in children and regular follow up are essential for good long-term visual outcomes. Younger children undergoing lensectomy have better visual outcomes but are at higher risk of glaucoma, but conversely children having surgery later are at higher risk of strabismus. 80 Non-surgical management may be appropriate with dilating drops for partial cataracts to increase light entering the eye. In galactosaemia cases, modification of diet and enzyme replacement may cause cataracts to stop progressing or even regress.83,84
Genetic testing
Genetic testing is a key investigation for congenital cataracts, and recent studies have shown molecular diagnostic rates between 50% and 90% for bilateral cases.3,85 Current practice involves using a targeted gene panel of cataract-related genes using next-generation sequencing technology, which has ⩾90% coverage. However, whole-genome sequencing (screening of all coding and non-coding intergenic and intragenic regions) has been estimated to increase diagnostics rates by a further 40%. 86 Genetic testing has been found to expedite diagnosis and provide a personalised clinical management plan, streamlining care pathways for patients, supporting informed genetic counselling and making informed decisions with regards to family planning. 5 Research focuses on integrating this evidence into clinical practice and establishing robust genotype–phenotype correlations to aid prognosis and to ensure systemic features are detected earlier to reduce comorbidities.87,88
Multidisciplinary care
Multidisciplinary management is essential to ensure full investigation and appropriate care pathways are established with paediatricians’ input, to exclude metabolic disorders, congenital infections and other syndromic and potentially life-threatening causes, especially while awaiting the results of genetic investigations.
Children require ongoing care within eye services where they will be seen regularly by ophthalmologists, orthoptists and optometrists to monitor the health and visual potential of the eye, and to ensure that any other amblyopic stimuli is minimised. In the UK, children and young people who are left with moderate reduction in their vision are referred to specialist advisory services for children with visual impairment in order to ensure that advice and support can be given to parents and to the educational services.
Assessment and clinical examination/investigation
An approach to assessment of children with cataract includes obtaining a history of the pregnancy and birth, with a focus on enquiring about maternal illness, drugs and medications in pregnancy, birth weight and neonatal events including jaundice treatment and clinical course. Subsequent history will be guided by the age of presentation, including current or past medical illness with particular attention to features of failure to thrive, developmental concerns and a thorough systems review. Family history and genogram should be obtained and investigations tailored to the history, age of presentation, gender and physical examination.
With a mobile and changing population, screening for congenital viral infections must be considered as there will be a small percentage of mothers who are not immune to rubella and thus at risk of becoming infected and passing on the infection to their unborn babies. Genetic testing for bilateral cases should be considered, whether through ophthalmic genetic specialists or clinical geneticists. 72
Vision, development and supporting families
Families of children with congenital cataract require information and support the first time they present to the eye clinic. Parents have concerns about their child’s prognosis and potential visual outcome, and how it will affect their future development and educational achievements. As children with sight impairment must optimise their vision, parents require guidance in the early years to know how to promote their child’s vision in order to maximise learning. Infants and children in the UK are referred to specialist teachers for children with visual impairment to provide crucial developmental guidance and support to families as the child moves through the toddler years into early nursery and statutory education. This support should go hand in hand with the paediatrician’s involvement, together with ophthalmology input with ongoing follow up in the children’s low-vision services.
Conclusion and future directions
Congenital cataract is a widely phenotypic heterogeneous disease with genetic and environmental causes to consider. Surgical management is the mainstay treatment for the cataract itself, but the clinical challenge is finding the cause. Many children presenting to health services with cataract with or without systemic features remain without a diagnosis. Genomic advances have accelerated the discovery of new cataract-causing mutations, and conditions associated with cataract continue to be identified. The ‘CAT-MAP’ database provides a comprehensive list of over 350 human and mouse genes and loci. 35 This includes at least 16 human loci where the underlying causative gene remains unknown, as well as a list of cataract genes shown to cause a cataract phenotype in mice. Although there are genotype–phenotype discrepancies between mice and humans, these lists suggest there are still many genes to be discovered. Recent research is directed towards providing high-quality evidence of the clinical utility of genetic testing to facilitate its provision and successful integration into clinical practice.85,88,89 Accurate diagnosis of inherited cataract is crucial for patients and their families as it facilitates individualised genetic counselling. It enables the multidisciplinary team to best support optimum child development during a critical period, empower families with a diagnosis and help them to plan for the future.
Footnotes
Author note
Mariya Moosajee is also affilited with The Francis Crick Institute, London, UK.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
This work was funded by the Wellcome Trust (Grant number 205174/Z/16/Z) and Leadiant Biosciences.