
Abstract
Background
Congenital heart disorders (CHD) are structural or functional heart diseases that are present at birth or found later in life. Conotruncal heart defects are one of the most common causes of symptomatic cyanotic cardiac disease. The 22q11.2 deletion syndrome is frequently linked to conotruncal abnormalities with varied presentation. The purpose of this research is to find out more about prevalence of 22q11.2 microdeletion and other genetic abnormalities in patients with conotruncal anomalies and its impact so as to formulate a genetic screening protocol.
Primary Objective
The main aim of this study was to analyse the frequency of 22q11.2 deletion in syndromic and nonsyndromic conotruncal anomalies.
Secondary Objectives
The secondary aim of this study was to determine the frequency of karyotypic or chromosomal abnormalities other than 22q11.2 deletion in patients with conotruncal anomalies. This study also aimed to compare the postoperative outcome in 22q11.2 deletion positive and negative cases of conotruncal anomalies.
Materials and Methods
This study was prospective observational study conducted in the Pediatric Cardiology Department of Star Hospital, Hyderabad, Telangana, India, over a period of two years. Genetics evaluation mainly included multiplex ligation-dependent probe amplification (MLPA) for 22q11.2 microdeletion and Karyotype. SALSA MLPA Probemix P064-C2 microdeletion syndromes-1B was used in this particular study to detect aberrant copy numbers of several chromosomal regions known to cause mental retardation syndromes.
Results and Discussion
This prospective observational study was conducted from December 2019 to November 2021. During this period, a total of 648 pediatric patients with cardiac anomalies requiring either surgery or intervention were admitted to hospital, of which 74 cases of conotruncal anomalies were included in this study. Median age at admission to hospital was 11 months, median weight was 7.6 kg (interquartile range [IQR]: 6.6–10), and median height/length was 70 cm (IQR: 67–80). Most common conotruncal anomaly in enrolled babies was tetralogy of Fallot (TOF) with good/fair pulmonary artery anatomy found in 30 (40%) cases. Extracardiac malformations were seen in 15 cases (20%). Among clinically syndromic cases, eight cases had abnormal genetic test (10.8%). None of the nonsyndromic cases had an abnormal genetic test. Frequency of 22q11.2 deletion in cases with conotruncal anomalies was 3 in 74 (4%). Frequency of other genetic syndromes was also seen in our study. In this study, there was no statistically significant difference in incidence of postoperative complications, death rate, or mean duration of intensive care unit (ICU) or hospital stay between genetic positive and negative cases.
Conclusion
Our research is the first population-based study of this magnitude using MLPA instead of fluorescence in situ hybridization (FISH) to examine the frequency of chromosome aberrations in children with conotruncal heart defects, especially 22q11.2 deletion syndrome and their impact on patient himself in terms of immediate outcomes after cardiac surgery, social, economic, and emotional impact on family and finally, on the whole healthcare system involved with the patient care.
Introduction
Congenital heart diseases (CHD) are structural or functional heart diseases which are present at birth or discovered at later age. CHD is known to exist 8-10 times per 1,000 live births and results from the interaction of multiple hereditary and/or nongenetic causes. 1 Identifiable genetic etiologies in syndromic CHD have been reported to be as high as 40%. 2 Conotruncal heart malformations are one of the most common causes of cyanotic cardiac diseases 3 and are embryologic malformations of the infundibulum (conus arteriosus) and great arteries (truncus arteriosus [TA]). Conotruncal defects include tetralogy of Fallot (TOF), transposition of great arteries (TGA), pulmonary atresia with ventricular septal defect (VSD), pulmonary valve agenesis, TA, double-outlet right ventricle (DORV), double-outlet left ventricle (DOLV), and interrupted aortic arch(IAA).4–12 Conotruncal defects are commonly found in patients with 22q11.2 deletion syndrome.5–8 The most frequent chromosomal microdeletion is the 22q11.2 deletion syndrome (also known as DiGeorge syndrome, velocardiofacial syndrome, and conotruncal anomaly syndrome) defined by heterozygous microdeletion of the 22q11.2 region of chromosome 22 and affects 1 in every 4,000 to 6,000 live births.13–16 Multiple fetal malformations and later-onset illnesses such as palate, gastrointestinal, and renal abnormalities, autoimmune disease, varying cognitive impairments, and psychiatric illness are all known features of the syndrome.9–14 Regardless of the magnitude of the deletions, the clinical manifestations of 22q11.2 deletion syndrome can range from modest-to-severe CHDs (77%), thymus aplasia/hypoplasia and immunological dysfunction (77%), palatal abnormalities (69%), learning difficulties (70%-90%), conotruncal facies, hypocalcemia (50%), and malformations of the genitourinary system (31%) 17 are the most common clinical characteristics of this syndrome. Karyotyping, chromosomal microarray, and fluorescence in situ hybridization (FISH) are frequently used to evaluate infants with CHD who also have additional severe abnormalities or dysmorphic characteristics. Researchers have attempted to determine the prevalence of 22q11.2 microdeletions in CHD patients with or without other symptoms of the syndrome in order to design a screening protocol for this disorder.18–20 Infants and children diagnosed early with 22q11.2 deletions may benefit from early intervention based on the evaluation and long-term care by multidisciplinary team. However, there are very few studies from India, reasons being late presentation, economic restraints, and limited resources. This study is aimed to ascertain the prevalence of 22q11.2 microdeletion and other genetic abnormalities in syndromic and nonsyndromic patients with conotruncal anomalies so as to formulate a genetic screening protocol best suited for our population.
Aim and Objectives
Primary Objective
The aim of this study was to analyse the frequency of 22q11.2 deletion in syndromic and nonsyndromic conotruncal anomalies.
Secondary Objectives
The following are the secondary aims of this study:
To determine the frequency of other karyotypic or chromosomal abnormalities other than 22q11.2 deletion in patients with conotruncal anomalies. To compare the postoperative outcome in 22q11.2 deletion positive and negative cases of conotruncal anomalies.
Materials and Methods
This study was prospective observational study conducted in the Pediatric Cardiology Department of Star Hospital, Hyderabad, Telangana, India, from December 2019 to November 2021. All cases of conotruncal anomalies admitted and evaluated during the study period were included. As we did not have a genetic department at our hospital, for patients’ convenience, genetic testing was done at the Department of Medical Genetics, NIMS (Nizam’s Institute of Medical Sciences) Hospital, Hyderabad, which has a very active genetic department. A total of 74 patients with various conotruncal anomalies underwent surgical correction in this period and were screened for deletion 22q11 and other genetic abnormalities. This study was started after receiving approval from the Institutional Scientific and Ethical Committee. Those with a conotruncal defect and any of the accompanying characteristics, such as dysmorphic facies, developmental delay, or extracardiac anomalies, were considered syndromic. All patients underwent a thorough cardiovascular examination, including two-dimensional echocardiography, electrocardiogram (ECG), and other necessary investigations such as cardiac catheterization, computed tomography angiography, or magnetic resonance imaging when indicated. Clinical genetics evaluation included a detailed history of presenting complaints, comprehensive family history, including consanguinity, anthropometric measurements, type of CHD, presence or absence of dysmorphism, extracardiac abnormalities, and psychomotor development. Parents were also counseled on the need for and implications of the patient’s genetic evaluation. The majority of the genetics examination consisted of:
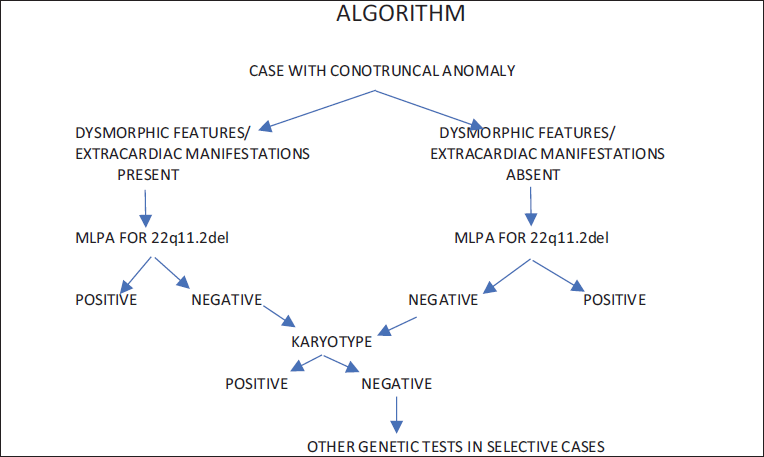
Multiplex ligation-dependent probe amplification (MLPA) for 22q11.2 microdeletion, which is the variation in the multiplex polymerase chain reaction that permits amplification of multiple targets with only a single primer pair. SALSA MLPA Probemix P064-C2 Microdeletion Syndromes-1B was used in this particular study to detect aberrant copy numbers of several chromosomal regions known to cause mental retardation syndromes. This probe mix has seven probes in the 22q11.2 region and can detect typical as well as atypical deletions. Karyotype is a procedure in which cytogeneticists image chromosomes to determine an individual’s chromosomal complement, including the number of chromosomes and any abnormalities. Karyotyping was done on all of the patients in this research to seek for chromosome 22 rearrangements in those who had a 22q11.2 deletion, and to look for numerical and structural abnormalities of other chromosomes in those who did not have a 22q11.2 deletion (Figure 1).
Statistical Analysis
Data were collected and spread in a Microsoft Excel sheet and were analysed using software SPSS version 23. The continuous data was measured by using mean ± SD or median (interquartile range) depending on the normality of distribution. Means were compared between two groups using independent t-tests. Medians were compared between two groups using the Mann–Whitney U test. Categorical data was expressed in proportions or frequencies and compared by using chi-squared or Fisher exact test. All tests used a significance level of P < .05.
Results
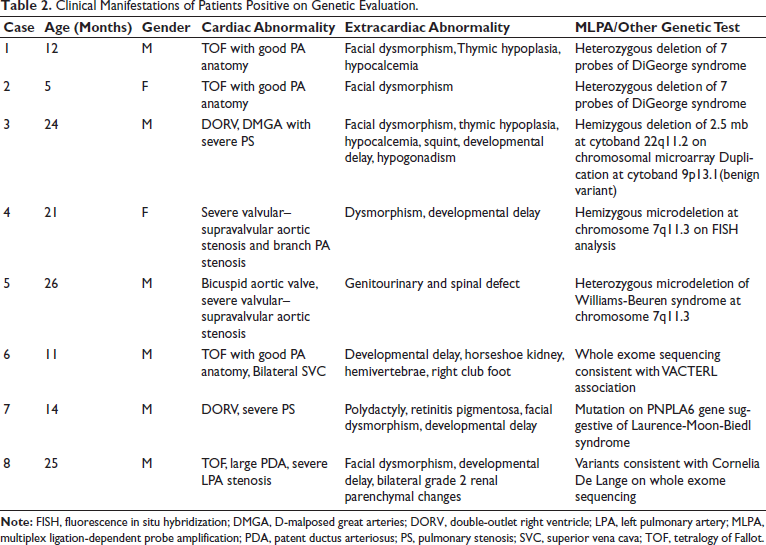
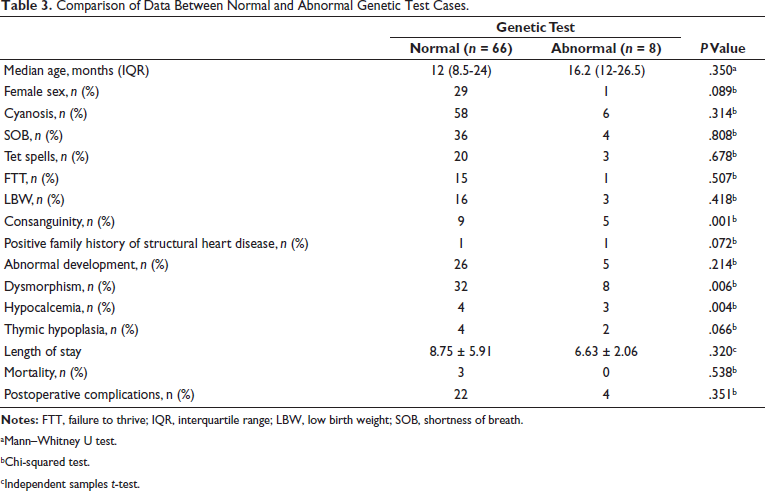
This prospective observational study lasted 24 months, during which 648 pediatric patients with cardiac anomalies requiring surgery or intervention were brought to the hospital, with 74 cases of conotruncal malformations being included in the study. Detailed demographic characteristics of enrolled patients are mentioned in Table 1. Most common conotruncal anomaly in enrolled babies was TOF, out of which TOF with good/fair pulmonary artery (PA) anatomy was found in 30 (40%) cases, and TOF with hypoplastic PAs was found in 6 (8%) cases. DORV associated with pulmonary stenosis (PS) with VSD was seen in another 13 (17%) cases. TA was diagnosed in six (8%) cases, and TGA was seen in another six (8%) cases. Pulmonary atresia with or without VSD was diagnosed in nine (12%) cases, three cases (4%) were diagnosed with left ventricular outflow anomalies and one with malaligned VSD with branch PA stenosis. In 15 cases, extracardiac abnormalities were discovered (20%). The most prevalent symptom in enrolled patients was cyanosis, which was seen in 64 (86%) cases, followed by increased work of breathing in 40 (54%) cases, Tet spells in 23 (31%) cases, and failure to thrive in 16 (21.6%) cases. During surgery, 41 cases (55.4%) were found to be syndromic, 31 (41.8%) to have developmental delay, seven instances to have hypocalcemia, and six cases to have hypoplastic/absent thymus. Among all the operated cases, three cases (4%) died in postoperative period. Twenty-six of the remaining 71 cases (35.1%) had one or more postoperative problems. Sinus tachycardia was the most common postoperative complication seen in six (23%) instances, followed by pericardial effusion in three cases and residual VSD in three more. Sepsis (3 cases), pulmonary arterial hypertension (2 cases), left ventricular dysfunction (2 cases), complete heart block (2 cases), arterial thrombosis (2 cases), seizures (2 cases), femoral artery stenosis (1 patient), acute kidney injury (1 patient), pulmonary bleed (1 patient), and pulmonary edema (1 patient) were among the other complications observed. MLPA/chromosomal microarray was abnormal in four cases, of which three cases were of 22q11.2 deletion syndrome (22q11.2DS), and one case was of Williams syndrome. For one case, 7q11.23 deletion (Williams syndrome) was diagnosed on FISH. Out of three cases in whom whole exome sequencing was done, one revealed variants pathgnomic of VACTERL association, another was diagnosed with Cornelia de Lange syndrome and one case of Laurence moon Biedl syndrome. None of the nonsyndromic cases had an abnormal genetic test. In our study, frequency of 22q11.2 deletion in cases with conotruncal anomalies was three in 74 (4%) cases, and some other genetic defects were also detected in some patients (Table 2). Among the conotruncal anomalies, consanguinity was common in those with an abnormal genetic test (25%) than those with normal genetics (18%), though the difference was not statistically significant. Cyanosis, shortness of breath, Tet spells, failure to thrive, and developmental difficulties had no statistically significant difference. Dysmorphism was more common in subjects with abnormal genetic tests, at 100% versus 49.2% in the normal genetic test group, with a significant P value of .026. Between the two groups, there was no statistically significant difference in the incidence of postoperative complications, death rate, or mean length of hospital stay (Table 3).
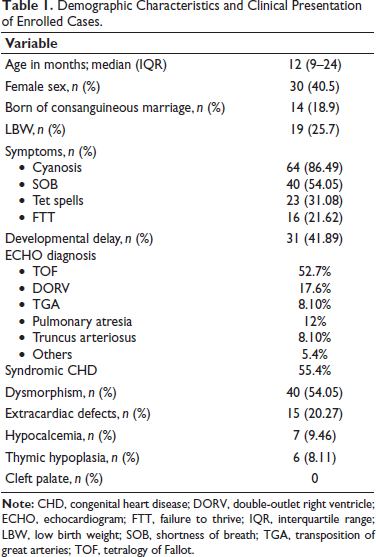
Demographic Characteristics and Clinical Presentation of Enrolled Cases.
Clinical Manifestations of Patients Positive on Genetic Evaluation.
Comparison of Data Between Normal and Abnormal Genetic Test Cases.
aMann–Whitney U test.
bChi-squared test.
cIndependent samples t-test.
Discussion
In our study, we discovered that eight of the clinically syndromic cases had abnormal genetic tests (10.8%) and incidence is comparable to already established data. There were no aberrant genetic results in any of the nonsyndromic patients which can guide us in restricting our genetic testing in syndromic CHDs only. In patients with conotruncal abnormalities, the frequency of 22q11.2 deletion was 3 in 74 cases (4%). Another Indian study by Halder et al. found that children with CHDs referred to a tertiary care center in north India had a prevalence of 6.16% (9 out of 146 patients) of 22q11.2 microdeletion, with all positive patients having extracardiac characteristics. 21 In addition to DiGeorge syndrome, we found the prevalence of other genetic abnormalities in our study. Additional genetic disorders other than 22q11.2 microdeletion were also found in patients with conotruncal abnormalities in a study by Lahiri et al. 22 All three cases with a positive 22q11.2DS on genetic examination had a clinical diagnosis of TOF physiology, two of which were TOF and one was DORV, VSD, and PS. It has long been known that anomalies on chromosome 22q11 are linked to TOF. 5 Furthermore, Pierpont et al. 23 found that children with TGA rarely have a 22q11 microdeletion, which is consistent with our findings. 24 Deletion 22q11 virtually always presents with one or more noncardiac anomalies of DG/VCFS. Dysmorphic faces was more commonly seen in cases with abnormal genetic test, 100% versus 49.2% in group with normal genetic test (P value: .026). (85.7%) had dysmorphic facies. Similarly, hypocalcemia was more commonly seen in group with abnormal genetic test, 33.3% versus 6.1% in group with normal genetic test (Table 3). It is similarly reported by Formin et al. 25 and others that characteristic facial features (conotruncal facies) followed by hypocalcemia are most common extracardiac manifestations of DiGeorge syndrome. Among the conotruncal anomalies, consanguinity was common in those with an abnormal genetic test (25%) than those with normal genetics (18%); however, the difference was not statistically significant (P value: .08). On the other hand, in this study, there was no statistically significant difference in incidence of postoperative complications, death rate or mean duration of intensive care unit (ICU) or hospital stay between two groups. It is contrary to what is reported by Lahiri et al., where there was significant difference in perioperative outcomes between syndromic and nonsyndromic pediatric patients who underwent surgical correction of CHD. Aggressive preoperative workup to rule out infections, identifying and managing other noncardiac issues before proceeding for cardiac surgery/intervention, delayed extubation with more intensive care, and vigilance in case of syndromic presentation in our hospital are some of the reasons of improved perioperative outcome except for prolonged ICU stay of syndromic cases in this study.
FISH, which uses a probe that maps to the LCR22A-LCR22B region, has been the most common technique used to diagnose a 22q11.2 deletion in the past. However, patients with minor or unusual deletions may be missed.21, 26–29 Ours was the first study in Indian settings to use MLPA instead of FISH. When compared to FISH, MLPA can identify patients with both typical and atypical deletions and is a more cost-effective and time-consuming method. SALSA MLPA Probemix P064-C2 Microdeletion Syndromes-1B was used in this particular study to detect aberrant copy numbers of several chromosomal regions known to cause mental retardation syndromes. In this work, we reported two cases with heterozygous deletion at 22q11.2 on MLPA and one case with heterozygous deletion in the 22q11.2 region on chromosomal microarray. Also, two cases of Williams syndrome were diagnosed one on MLPA, other on FISH (done outside) with heterozygous deletion at 7q11. Other syndromes in this study were diagnosed on whole exome sequencing as advised by the geneticist in view of their clinical spectrum and presentation. However, targeted gene sequencing could also be done, but there were chances of missing out on rare genetic disorders. The management of 22q11.2DS necessitates a personalized, interdisciplinary, and coordinated care strategy that considers the patient’s other characteristics. The International 22q11.2DS Consortium was founded in 2006 with the purpose of producing management protocols and standards for people with 22q11.2DS, both children and adults (International 22q11.2DS). The mainstay of treatment protocol is early recognition, comprehensive clinical workup, including genetic evaluation, and predicting the issues that may arise in a syndromic child who has not yet undergone cardiac surgery. Hypocalcemia, immunological depression, vasomotor instability, bronchospasm, and airway hemorrhage should all be avoided with special perioperative care.23, 30–34 Prevalence of 22q11.2DS and other genetic abnormalities in patients with conotruncal anomalies has been estimated in this study in an attempt to establish a screening protocol for this condition, with or without other features of the syndrome. Prenatal counseling for future pregnancies is also required, although there is only 1%-2% chance of recurrence.
Conclusion
Our research is the first population-based study of this magnitude using MLPA to examine the frequency of chromosome aberrations in children with conotruncal heart defects, especially 22q11.2 deletion syndrome and their impact on patient himself in terms of immediate outcomes after cardiac surgery. Screening with baseline genetic tests should be restricted to syndromic conotruncal cases only as none of nonsyndromic had abnormal genetic results.
Limitations
The study is prospective observational in nature and subject to limitations inherent to observational investigations. Only basic genetic workup was done in all, and advanced genetic testing was limited to a few cases because of economic constraints, even if there was strong suspicion of underlying genetic abnormality. Many confounding variables like availability of resources, socioeconomic status of the patients, intraoperative and postoperative management affect the outcomes. Well-designed prospective studies focused on long-term surgical outcomes in syndromic conotruncal anomalies and recurrence in future pregnancies should be conducted.
Author Contributions
The article was conceived by Dr. Radhika. All other authors contributed to the collection of photographs, videos, and the preparation of the manuscript. All authors participated in the revision and critical review of the article.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This study is a retrospective case review. The need for ethical approval and informed consent was waived by the Ethics Committee of Star Hospital.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Patient Consent
No photographs or identifying information of individual patients have been included in the manuscript.