
Abstract
Cardiac amyloidosis wild type (ATTR-CAwt) is often the underdiagnosed cause of heart failure with preserved ejection fraction (HFpEF) and has a high mortality rate. There is usually a long delay between the appearance of clinical signs and the diagnosis of ATTR-CAwt but a short duration between diagnosis and death. ATTR-CAwt was associated with significant clinical arrhythmias. We report a case of ATTR-CAwt, the process of its diagnosis, and the associated clinical arrhythmias and their management. We also reviewed the literature of this underdiagnosed and potentially fatal condition and the current existing therapies.
Case Report
A 79-year-old African American male with a history of hypertension, hyperlipidemia, and chronic asthma was evaluated in our office. The patient complained of chest tightness, difficulty in breathing, and bilateral lower extremity swelling for several months (approximately 6 months) which was progressively worsening. The patient also complained of occasional, positional lightheadedness. Review of systems was positive for bilateral upper extremity wrist pain/carpal tunnel syndrome. The patient was treated with multiple courses of antibiotics for bronchitis for the last 6 to 9 months. He was not a current smoker (quit 20 years ago) and consumes 1 to 2 glasses of wine per day. Family history was significant for “mother with fluid around the heart” but with unknown diagnosis.
Examination demonstrated that his jugular venous pulse elevated to 10 cm H2O, normal S1 and S2 with nondisplaced apex (point of maximum impulse), and decreased breath sounds at bilateral lung bases with rales in lower lung zones. There was occasional expiratory wheezing. There was 2+ pitting pedal edema of bilateral lower extremities.
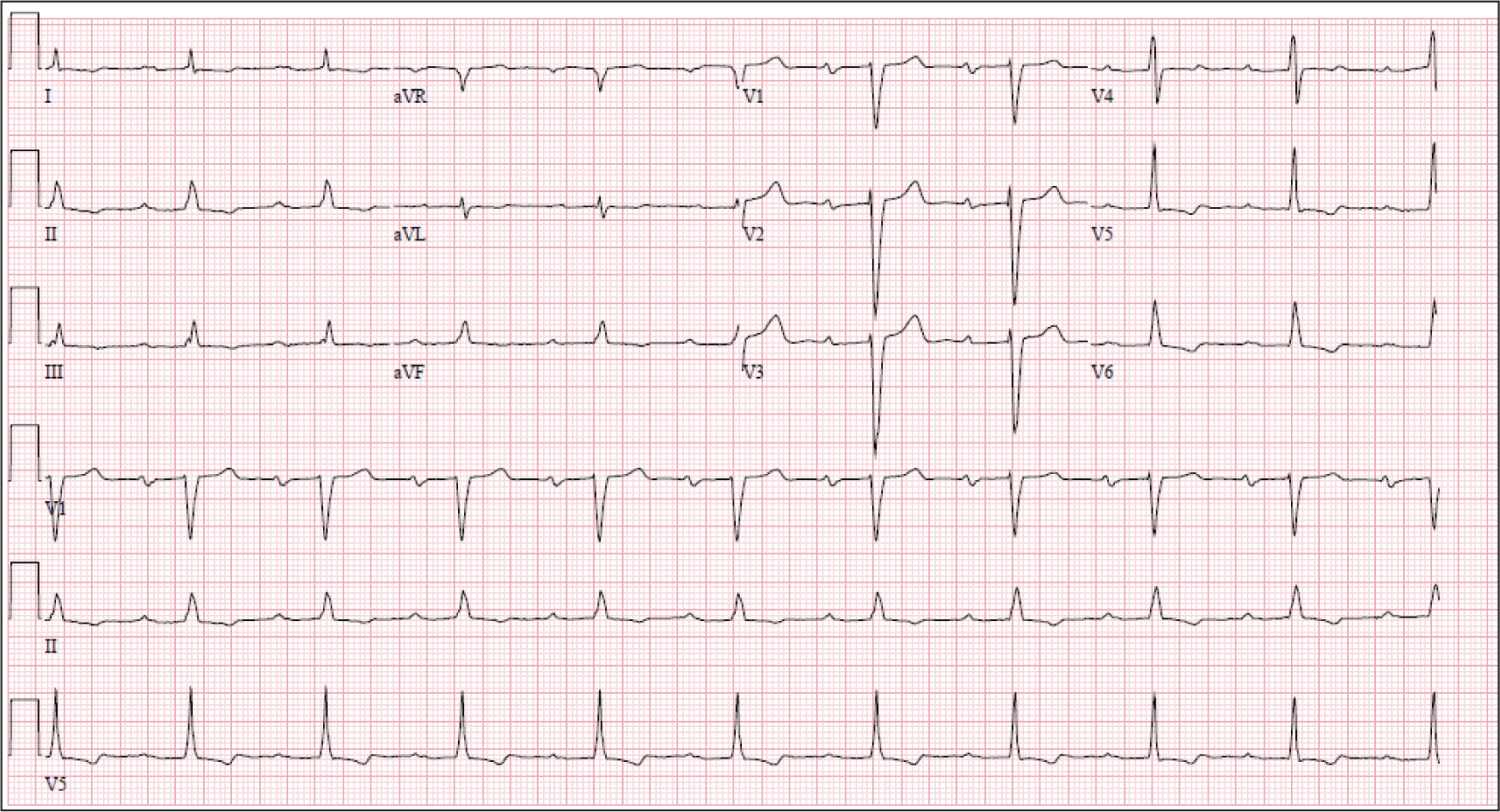
Baseline electrocardiogram (Figure 1) in the office demonstrated sinus rhythm (SR) with first degree atrioventricular (AV) block and QRS with intraventricular conduction defect. There were T-wave inversion in limb leads II, III, aVF, and precordial leads V4 to V6.
Electrocardiogram showed Sinus rhythm (SR) with first degree AV block; QRS with Intraventricular conduction defect (IVCD); T-wave inversions (TWI) in inferior and lateral precordial leads.
Given episodic chest tightness and symptoms of heart failure, patient was hospitalized and underwent blood work. Lab tests demonstrated pro-B-type natriuretic peptide (pro-BNP) of 5,500 pg/mL, troponin of 0.10 to 0.12 ng/mL (normal <0.02 ng/mL), and creatinine of 1.6 mg/dL (normal 0.5-1.2 mg/dL). He was started on a diuretic.
In the hospital, he underwent cardiac workup as follows:
Cardiac echocardiogram demonstrated moderate concentric left ventricle (LV) hypertrophy with moderate LV systolic dysfunction with left ventricular ejection fraction (LVEF) of 45%. There was mild right ventricle (RV) dilation with probably normal RV function. There was moderate to severe left atrial (LA) enlargement and moderate right atrial enlargement. There was mild tricuspid regurgitation with RVSP ~25 mm Hg.
Cardiac catheterization was performed and showed no obstructive epicardial coronary artery disease and LVEF of 45%.
As workup for nonischemic cardiomyopathy, he underwent cardiac magnetic resonance imaging (MRI) (Figure 2), which demonstrated LV-end diastolic volume of 75 cc/m 2 and mild global LV hypokinesis with LVEF of 45%. There was asymmetric septal and inferoseptal LV hypertrophy with maximum thickness at basal septum of 19 mm and RV with normal thickness and borderline hypokinesis. There was biatrial enlargement. Delayed contrast enhancement showed a predominantly LV mid-wall enhancement of septum, inferior, and inferolateral walls, and transmural enhancement of the basal inferior, inferoseptal, and inferolateral LV walls. Small pericardial and pleural effusions were noted. These findings were consistent with infiltrative nonischemic cardiomyopathy.
Cardiac magnetic resonance imaging (MRI) delayed contrast enhancement LV short axis (a) and long axis views (b) showed a predominantly LV mid wall enhancement of septum, inferior and inferolateral walls and transmural enhancement of the basal inferior, infero-septal and infero-lateral LV walls.
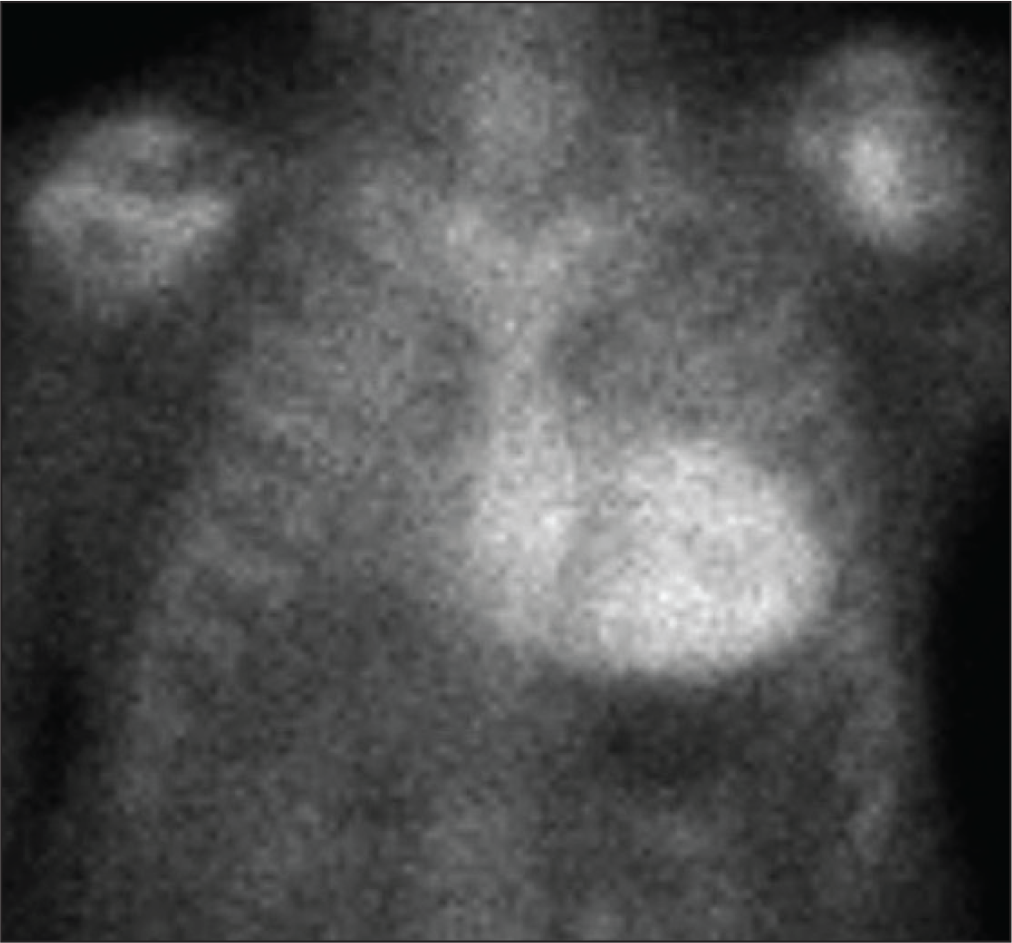
Because of the above findings and due to suspicion of the infiltrative cardiomyopathy including amyloid cardiomyopathy, patient then underwent technetium pyrophosphate nuclear scan (99mTc-PyP). There was diffuse uptake present throughout the LV myocardial segments with heart to contralateral ratio at 1 h elevated at 2.4, and also elevated at 3 h at 2.1 (Figure 3). These findings were strongly suggestive of transthyretin cardiac amyloidosis of the wild type (ATTR-CAwt).
He underwent urine protein electrophoresis (UPEP) and serum protein electrophoresis (SPEP) with immunofixation. UPEP and SPEP showed no free light chains and no monoclonal gammopathy. He also underwent bone marrow biopsy and there was near normocellular bone marrow and no cytogenetic evidence of abnormal clone. Both abdominal fat skin biopsy and bone marrow biopsies had no evidence of amyloid deposition. All these findings ruled out AL amyloid.
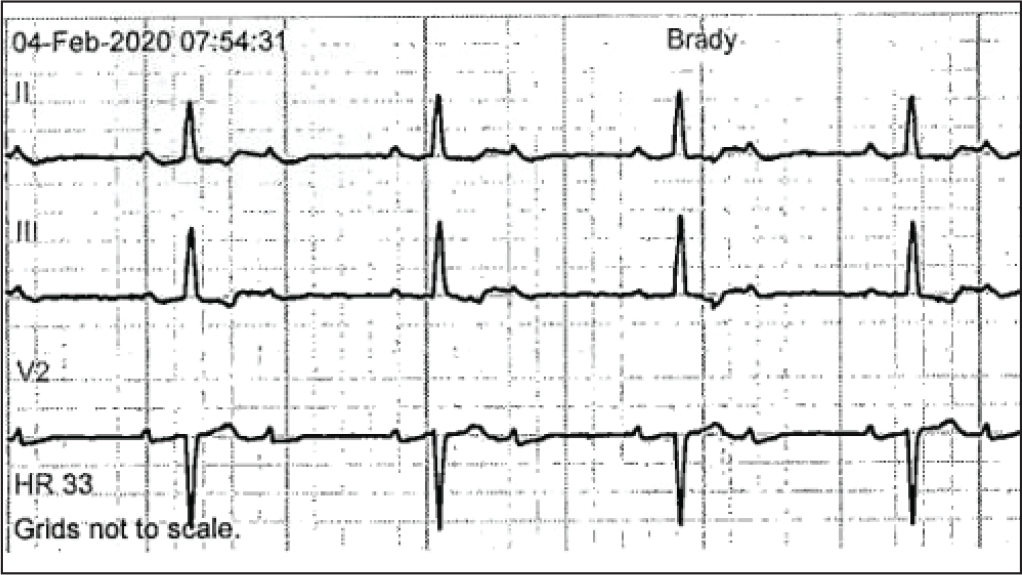
While being monitored in the hospital on telemetry unit, patient had episodes of significant bradycardia with high-grade Mobitz type 2:1 AV block with V rate of 30 s (Figure 4).
He then underwent electrophysiology study which demonstrated prolonged baseline His bundle-ventricular (HV) interval of 84 to 92 ms. The atrioventricular block cycle length was prolonged at 650 ms. There was no ventriculoatrial conduction at pacing cycle length of 700 ms. There were no inducible ventricular arrhythmias (VAs) with standard pacing protocol. He then underwent implantation of a dual chamber pacemaker implantation.
Technetium pyrophosphate nuclear scan (99mTc-PyP) showed diffuse uptake present throughout the LV myocardial segments with Heart to contralateral ratio at 1 h elevated at 2.4; and also elevated at 3 h at 2.1.
Telemetry monitor strips showed significant bradycardia with high grade Mobitz type 2:1 AV block with V rate of 30 s.
Subsequently, the patient was diagnosed with atrial fibrillation (AF) on pacemaker device checks. He had a CHADS-VASc score of 4. He was started on anticoagulation with apixaban for stroke prevention of AF. He was initially rate-controlled with beta blocker and was later started on amiodarone for rhythm control.
Genetic testing was performed and his amyloidosis was confirmed to be the wild type (ATTR-CA wt). He was started on Tafamidis 61 mg daily (Trade name: Vyndamax-Pfizer pharma) by the heart failure team at our hospital and he tolerated the medicine well. He has now completed 3 months of therapy with no further congestive heart failure exacerbations or hospitalizations for heart failure.
Discussion
Amyloidosis is a disease associated with misfolded proteins that form amyloid fibrils and deposit in an organ, disrupting the organ’s structure and function. In the case of cardiac amyloidosis, protein deposits within the myocardial tissue. More than 30 proteins can form amyloid in vivo, but cardiac amyloidosis is predominantly caused by 2 precursor proteins—either immunoglobulin light chain (AL-CA) or transthyretin (ATTR-CA).1, 2 ATTR comes in 2 forms, a hereditary form (hATTR) or an age-related wild-type form (ATTRwt). ATTR wt, in which amyloid from normal TTR protein is deposited over a period of decades, has a strong male predilection and patients typically present between the ages of 60 and 95 years.
Because of its late presentation, ATTRwt is also referred to as senile amyloidosis.
Diagnosis of ATTR-CA
There is usually a significant delay in the accurate diagnosis of cardiac amyloidosis. There are several reasons. One major reason is the physician perception that cardiac amyloidosis is an extremely rare disease. ATTRwt-CA is not that rare, particularly when we focus on specific patient populations and disease characteristics. Approximately, 13% of patients who have ATTRwt-CA are hospitalized for HFpEF. 3 About 16% of patients with severe aortic stenosis (AS) and aortic valve replacement (AVR) have ATTRwt-CA. 4 In this series, 1 in 5 men undergoing a transcatheter AVR had ATTRwt amyloidosis and it is even more common in those with a low-flow, low-gradient AS phenotype. The Val122Ile mutation, an inherited form of hATTR, is observed in 3.4% of Black individuals, which translates to about 1.5 million people in the United States who are allele carriers.5- 7 Depending on penetrance, more than 200,000 Black individuals may manifest hATTR-CA. These data indicate that the condition is not rare.
As per a famous medical saying, “You will never make a diagnosis that doesn’t enter into your differential.” Earliest manifestations of ATTR-CA could be noncardiac. A history of carpal tunnel syndrome (especially if bilateral) is one of the earliest indicators of ATTR-CA and can precede clinical heart failure by several years. Lumbar spinal stenosis and ruptured distal biceps tendon are also early clinical manifestations of ATTR-CA.
A disproportionately low QRS voltage on electrocardiogram to LV thickness or LV mass ratio is an earlier “signal” and more sensitive for identifying patients with potential ATTR-CA.8, 9
No biomarker is currently diagnostic of ATTR-CA. However, a combination of elevated troponin and an NT-proBNP level that is disproportionate to the degree of heart failure should prompt diagnostic evaluation. 10
Echocardiography can reveal several clinical clues raising suspicion for ATTR-CA, including thickening of the LV wall (≥12 mm) and interatrial septum. 11 Apical sparing of global longitudinal strain relative to the LV base and mid-ventricle is another clue, referred to as an “ice cream cone with a cherry on top.” CMR imaging exhibiting diffuse delayed gadolinium enhancement is also suggestive of cardiac amyloidosis.
Be suspicious of ATTR-CA when patients with HFpEF frequently experience hypotension or orthostasis and become intolerant of antihypertensive medications. These signs could be caused by amyloidosis-reducing cardiac output and cause hypotension. 12
The diagnosis of ATTR-CA begins with a heightened clinical suspicion followed by the exclusion of AL-CA. AL-CA can mimic the echocardiographic findings of ATTR-CA and is a common cause of false-positive nuclear scintigraphy scans. AL-CA is excluded by screening for monoclonal gammopathy with serum-free light chains and UPEP and SPEP with immunofixation. 13 A negative finding on these 3 tests sufficiently excludes AL-CA as a diagnosis.
Both planar and single-photon emission computed tomography (SPECT) imaging can noninvasively diagnose ATTR-CA (without endomyocardial biopsy) with high sensitivity and specificity. 14 Only the following nuclear radiotracer should be used; 99mTc-PYP in the United States and 99mTc-DPD or 99mTc-HMDP in Europe. Images should be obtained 1 to 3 h after the isotope is injected. 15 A grading system comparing myocardial uptake of 99mTc-PYP in the heart vs. the rib bone is used: grade 0 (no cardiac uptake), grade 1 (cardiac less than rib), grade 2 (cardiac equal to rib), and grade 3 (cardiac greater than rib). Grades 2 and 3 are strongly suggestive of ATTR-CA. The quantitative approach compares circular regions over the heart (H) and contralateral lung (CL) fields. An H/CL uptake ratio ≥1.5 at 1 h or ≥1.3 at 3 h after injection is also strongly suggestive of ATTR-CA. 16
If all the following criteria are met, a definitive diagnosis of ATTR-CA can be made in the absence of an endomyocardial biopsy:
Echocardiographic and/or CMR findings suggestive of cardiac amyloidosis.
The presence of ≥grade 2 uptake by 99mTc-PYP that is confirmed by SPECT imaging.
The absence of a monoclonal gammopathy by serum-free light chain assay and serum and urine immunofixation electrophoresis.
Therapies
Chemotherapy which is useful for AL-CA has no role in ATTR. Among the disease specific interventions for ATTR-CA, liver transplantation does remove the mutant amyloidogenic TTR in ATTRm; however, in wild type ATTR-CA, the precursor protein is native TTR and liver transplantation is thus not beneficial. Novel disease-specific therapies directed at underlying protein misfolding in ATTR-CA are constantly evolving. Tafamidis is a drug that binds to and stabilizes transthyretin, thereby preventing transthyretin tetramer dissociation, the rate-limiting step in transthyretin amyloid deposition. 17 Multicenter randomized ATTR-ACT trial demonstrated that in patients with ATTR-CA, Tafamidis was associated with reductions in all-cause mortality (Absolute risk reduction [ARR] 13.4%) and cardiovascular-related hospitalizations (ARR 22%) and reduced the decline in functional capacity and quality of life. 18 Drugs inhibiting transthyretin messenger RNA (mRNA) and protein synthesis is another strategy which is under development and among them Patisiran (small interfering RNA that specifically targets and silences transthyretin mRNA) and Inotersen (single stranded oligonucleotide that binds to mRNA causing degradation and reduced expression) have shown to be beneficial in neuropathy19, 20; however, cardiac data is limited. Another proposed strategy is increased clearance of amyloid fibrils; however, it largely remains investigational. Currently, Tafamidis is the only approved treatment for ATTR-AC in the United States, while Patisiran, Inotersen, AG-10 (TTR stabilizer), and Ab-A (mAb promoting clearance and degradation of aggregated TTR) are being evaluated in clinical or preclinical studies for ATTR-AC. 21
Arrhythmic Manifestations of ATTR-CA and Their Management
Patients with cardiac amyloidosis also develop progressive conduction disease. AV block or sinus node dysfunction in an older patient with increased LV wall thickness should raise suspicion for cardiac amyloidosis. Electrophysiologic studies show an abnormal HV interval in most patients. Prolongation of the HV interval with preserved QRS duration is well recognized. This may represent diffuse amyloid infiltration of the bundles, creating equal delays in both the right and left branches, yielding a disproportionately narrow QRS. 22 Approximately, 45% of ATTR-CAwt patients require pacemaker implants.
Atrial arrhythmia, usually AF, is very prevalent (~44%) in ATTR-CA wt. AF is poorly tolerated due to rapid ventricular rates. Beta blockers and Calcium channel blockers are not well tolerated due to hypotension and their negative inotropic effects. Low doses of digoxin are usually well tolerated. 23 Amiodarone provides a good option for AF control and is fairly well tolerated. Cardiac ablation is tolerated with limited success. 24 Despite acute termination and restoration of SR with ablation, the recurrence rate is very high. In one series, 17 the recurrence rate at 1 year was 83% for CA patients compared with 25% in the non-CA, persistent AF patient group. The high rate of recurrence in ATTR-CA wt is possibly due to associated diffuse biatrial myopathy. 25
Cardiac amyloidosis has been associated with a high risk of intracardiac thrombus, stroke, and systemic embolism. Pathophysiologic mechanism is amyloid infiltration involving both the right and left atria leading to reduced atrial contractility, blood stasis, endothelial dysfunction, and relative hypercoagulability. There is no association between the CHA2DS2-VASc score and the presence of LA appendage thrombus on transesophageal echocardiogram. Some leading experts suggest that all patients with AF and cardiac amyloidosis be offered oral anticoagulation regardless of CHA2DS2-VASc score. 26
One-half (50%) of patients with cardiac amyloid die suddenly. Guidelines do not strongly support implantable cardioverter-defibrillator (ICD) placement for both primary and secondary prevention in cardiac amyloid currently due to the following reasons:
The most common cause of sudden death has been secondary to electromechanical dissociation, resulting in pulseless electrical activity rather than lethal VAs.
Higher defibrillation thresholds.
Historically poor prognosis and life expectancy.
ICD therapy was not associated with improved mortality in follow-up.
The 2017 American Heart Association/ACC/HRS guideline for management of patients with VAs and the prevention of sudden cardiac death recommends individualized decision-making for both primary and secondary prevention ICD. 27
Conclusion
ATTR-CA is not a rare condition and it is essential to have high index of suspicion. ATTR-CA is commonly misdiagnosed as HFpEF of unknown cause, hypertrophic cardiomyopathy, hypertensive heart disease, or degenerative AS.
A history of bilateral carpal tunnel, cardiac symptoms accompanied by autonomic dysfunction, and unexplained increased LV wall thickness can be clues.
Disproportion between LV wall thickness and QRS voltages on electrocardiogram is the hallmark feature of cardiac amyloidosis.
Atrial and ventricular arrhythmias and conduction disease are common in cardiac amyloidosis.
AF may be difficult to rate control and carries a high risk of stroke. Anticoagulation is recommended in these patients with AF and cardiac amyloidosis regardless of the CHA2DS2-VASc score.
Implantable cardioverter-defibrillator placement for prevention of sudden cardiac death is controversial.
New therapies are likely to change prognosis and therefore, further studies are needed.
Footnotes
Declaration of Conflicting Interests
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.